| 18 |
Metabolism of reactive species |
| 18.1 |
Introduction |
Low-molecular reactive species such as superoxide (O2•−), hydrogen peroxide (H2O2), nitric oxide (•NO), and peroxynitrite (HOONO) are of great importance in biology. Because of their high reactivity, they are an unruly bunch: unlike the sedate, well-behaved metabolites of regular pathways such as glycolysis, reactive species don’t wait to be chaperoned along the beaten path by some friendly neighborhood enzymes; instead, they tend to react on their own at every opportunity, whether the outcome be useful or destructive. In doing so, they create surprising links between pathways that we don’t usually think of as connected; for example, uric acid and bilirubin, the respective end products of purine and heme degradation, may scavenge reactive oxygen species that were produced in mitochondrial respiration or through photosensitization by porphyrins in the skin.
The lion’s share of reactive species arises as by-products of regular metabolic pathways, particularly those that involve oxygen utilization and transport. However, they are also generated on purpose for transmitting signals within and between cells, or for killing infectious microbes. This latter function clearly shows the ability of reactive species to inflict serious cellular damage, and indeed our own body cells dedicate significant resources to preventing such damage to themselves. Consider for example glutathione, which is an important scavenger of reactive species: in liver cells, it is present at 7-8 mM [144] and thus more abundant than even ATP, which is the most common cosubstrate in the realm of purely enzyme-controlled metabolic cycles. In fact, the metabolic burden of defense against reactive species, particularly those derived from oxygen, is such that many microbial species forgo it altogether and confine themselves to purely anaerobic lifestyles and habitats.
Reactive species have already come up in several preceding chapters, for example in connection with LDL oxidation (see section 11.6.9) and with NADPH consumption (see section 9.3.4). Here, we will examine this somewhat complex and elusive subject in a more systematic manner. In keeping with the overall scope of these notes, we will focus on reactions and pathways that are relevant to human metabolism.
| 18.1.1 |
Reactive species in the human body: examples |
| Reactive species | Origin | Function or effect |
| O2•− | respiratory chain | byproduct |
| •OH | ionizing radiation; Fenton reaction | DNA damage, lipid peroxidation (cell membranes, LDL) |
| H2O2 | phagocytes | killing of microbes |
| thyroid peroxidase | reaction intermediate | |
| superoxide dismutase | detoxification intermediate | |
| HOONO | phagocytes | killing of microbes |
| singlet oxygen | photosensitization in porphyrias | skin damage |
| N-acetyl-p-quinone-imine (NAPQI) | metabolite of acetaminophen | drug toxicity |
| R–S• | secondary radical | detoxification intermediate |
The examples in this table make it clear that reactive species are diverse with respect to both chemical nature and biochemical context. We can observe that many reactive species, but not all, are radicals; and moreover that many, but not all, contain oxygen. Those that do are commonly referred to as reactive oxygen species, or ROS for short. The term reactive nitrogen species is analogous; peroxynitrite (HOONO), nitric oxide (•NO), and nitrogen dioxide (•NO2) are included in both groups.
The examples illustrate the scope of the subject, but they do not exhaust it. For example, H2O2 also occurs as a metabolic byproduct with xanthine oxidase (see slide 16.5.3), while •OH and singlet oxygen are also formed on purpose in phagocytes. Moreover, reactive species will often react and give rise to different ones before being scavenged; a cell that is trying to rein them in is thus faced with a veritable bag of fleas.
| 18.1.2 |
Do reactive species really matter in a class on metabolism? |
- Reactive species are intermediates or byproducts of metabolic reactions
- Reactive species participate in the development of atherosclerosis and other metabolic diseases
- Metabolites and enzymes that scavenge radicals are highly abundant—e.g. glutathione (7-8 mM in liver cells), peroxiredoxins (1% of cellular protein)
We have already considered the roles of ROS in the oxidation of LDL (see slide 11.6.9), in the destruction of red blood cells in glucose 6-phosphate dehydrogenase deficiency (section 9.4), and in skin damage in porphyrias (section 17.3). Thus, if we want to properly understand the connection of metabolism to health and disease, we must take reactive species into account.105
| 18.2 |
Chemistry of reactive species: some basic concepts |
While this is not a chemistry text, a brief survey of the chemistry of radicals and other reactive species may be useful.
| 18.2.1 |
Reactive species and ionizing radiation |
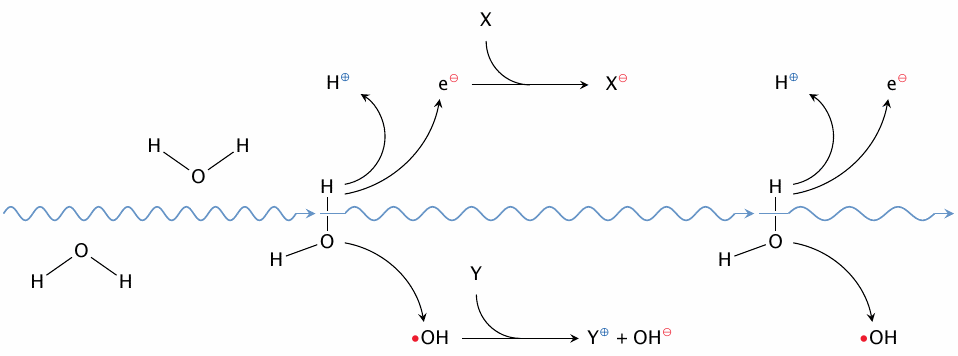
Ionizing radiation provides a straightforward source of radicals, and indeed its biological effects are mediated by those radicals.106 While the various types of radiation differ in the nature of the energy-rich particles—the picture is intended to illustrate a γ-particle, that is, a very high-energy photon—they all interact with human tissues by knocking free electrons.107 The loss of an electron from a molecule will often result in bond breakage. While radiation particles don’t really care what particular bonds or molecules they break, most often they will split water, simply because it is so abundant. As shown in the illustration, this will produce a hydroxyl radical as well as a free electron. Both will in turn react with other targets in the cell to produce a multitude of secondary ions and radicals—often simultaneously, that is, the reaction products may be radical anions and radical cations.
| 18.2.2 |
How toxic are •OH radicals? |
- The LD50 of γ-radiation is 5 Gray (Gy) = 5 J/kg
- The main effect of γ-rays is to break up H2O into H+, e−, and •OH
- The bond dissociation energy for the first bond in water is 500 kJ/mol, and that for ionizing the resulting hydrogen atom is 218 kJ/mol
- ⇒ at most 7 µmol/kg of •OH is produced by one LD50 of γ-rays
Ionizing radiation also gives us insight into the toxicity of reactive species. We can work out an upper limit for the concentration of hydroxyl radicals (•OH) from the lethal dosage of γ-irradiation.
The dosage of γ-radiation can be measured in units of energy deposited per kg of human tissue—ultimately in the form of heat, but initially mostly in the form of bond breakage. Even assuming that all of the absorbed energy goes into breaking up water, an LD50 of γ-radiation can produce no more than 7 µM of •OH.
You might now wonder what the point may be of keeping millimolar amounts of radical scavengers around, since they fail to cope with micromolar concentrations of •OH. It turns out that •OH is somewhat unique in this regard; it reacts very fast and indiscriminately with just about any biomolecule, which collectively exceed the radical scavengers in abundance. However, radical scavengers do work much better with other, less reactive radicals.
| 18.2.3 |
Reactions of radicals with each other and with non-radicals |
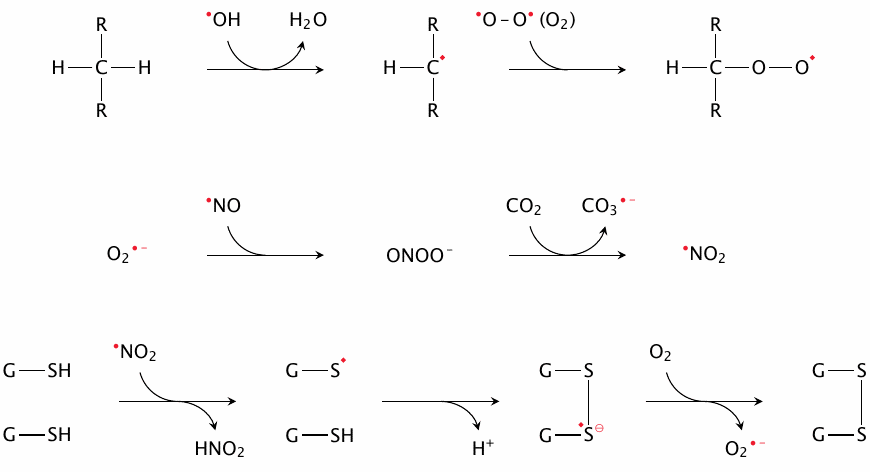
The top row shows the abstraction of hydrogen from carbon by •OH, which gives a carbon-centered radical. The latter is likely to recombine with another radical, often molecular oxygen, which is a diradical. Both of these reactions are important in lipid peroxidation (see section 18.5). Hydrogen can also be abstracted by other radicals and from other functional groups; for example, both ascorbate and α-tocopherol scavenge radicals by giving up hydrogen atoms that are part of their own hydroxyl groups, thus turning into radicals themselves (see section 18.7).
The middle row shows the recombination of O2•− and •NO into peroxynitrite. The latter, while not itself a radical, is highly reactive nonetheless and can give rise to various secondary radicals. Among the ones shown here, •NO2 tends to react with tyrosine residues or with thiol groups.
When •NO2, CO3•− or some other radical reacts with the thiol group of glutathione (G–SH, bottom row), a thiyl radical (G–S•) results that can react in turn with another glutathione molecule, forming a disulfide radical anion. The latter can offload its surplus electron to O2, giving O2•−. This reaction sequence is one of central pathways of radical scavenging—it serves to convert a multitude of different radical species to a single one (superoxide), which can then be disposed of in an an orderly manner by superoxide dismutase (see slide 18.7.3)
| 18.2.4 |
The reaction of H2O2 with thiol groups |
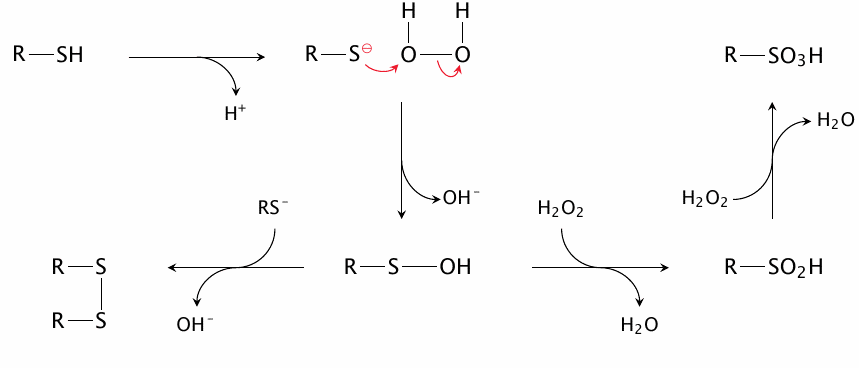
This reaction involves sulfhydryl groups in the deprotonated state; therefore, the pKa value of a given thiol is a key determinant of its reactivity towards H2O2 (and also towards alkyl peroxides). In contrast, thiol groups may react with radicals in their undissociated forms (see previous slide). Therefore, glutathione, whose pKa is 9.4 [147], reacts readily with radicals but fairly slowly with H2O2. Some protein thiol groups have significantly lower pKa values than glutathione and thus react faster with H2O2 (see slide 18.2.8).
The first equivalent of H2O2 converts a thiol group to sulfenic acid (R–SOH), which can either react with a second thiol group to form a disulfide, or with additional H2O2 to yield sulfinic acid (R–SO2H) and finally sulfonic acid (R–SO3H). Disulfide formation is reversible, while oxidation to sulfinic acid and beyond is not; thus, if a protein thiol that has been oxidized to a sulfenic acid group is to be preserved, disulfide formation must get there first. The second thiol group may be part of the protein itself, of glutathione, or of another molecule. Reducing enzymes such as thioredoxins and glutaredoxins can subsequently reduce the disulfide and restore the free thiol group (see section 18.7).
| 18.2.5 |
Radical reactions with transition metals |
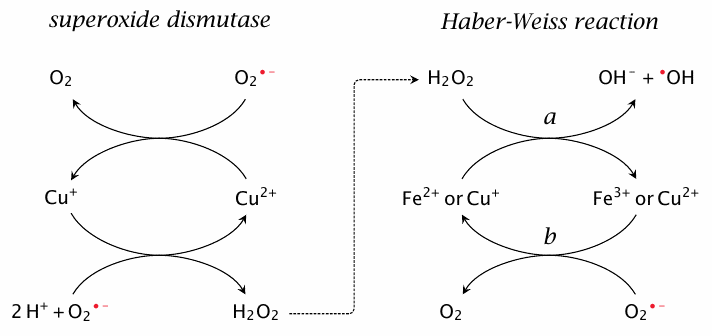
Both radicals and transition metal ions have at least one singly occupied orbital that may easily give up its electron or accept another. It is thus understandable that transition metals themselves behave quite similarly to radicals, and also that reactions between transition metals and radicals may generate other reactive species. What is more, such reactions can take the form of catalytic cycles that may produce more potent radicals (•OH) from less potent ones (O2•−).
The reaction sequence on the right (a and b) is known as the Haber-Weiss reaction, and step a—the decomposition of H2O2 to OH− and •OH by reduced iron or copper—as the Fenton reaction. It involves free copper or iron ions, which in vivo are normally very scarce. However, note that the reduced metal ions consumed by the Fenton reaction can be regenerated by O2•− and thus function in a catalytic manner. Furthermore, free heme and some other bound forms of iron may also facilitate this reaction. Happily, there are several pathways that scavenge H2O2 and thereby usually prevent •OH generation (see below).
The reactions shown on the left occur inside the active site of superoxide dismutase. This enzyme permits the formation of H2O2 but manages to prevent its further decomposition via Fenton chemistry. We will consider how this works in section 18.7.3.
| 18.2.6 |
Fenton-like radical formation by transition metals |
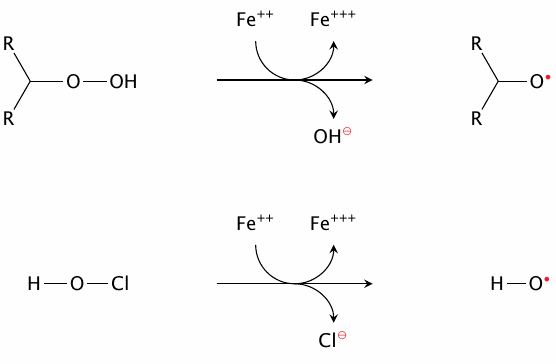
Transition metals may react similarly with compounds other than H2O2; for example, fatty acyl hydroperoxides maybe activated to alkoxyl radicals, and HOCl may be converted to •OH and Cl−. Moreover, reducing agents other than O2•− may reduce oxidized transition metals and thereby support repeated rounds of Fenton chemistry. Thus, overall, transition metals play a major role in the generation and turnover of reactive species.
| 18.2.7 |
Diffusion distances of selected reactive species |
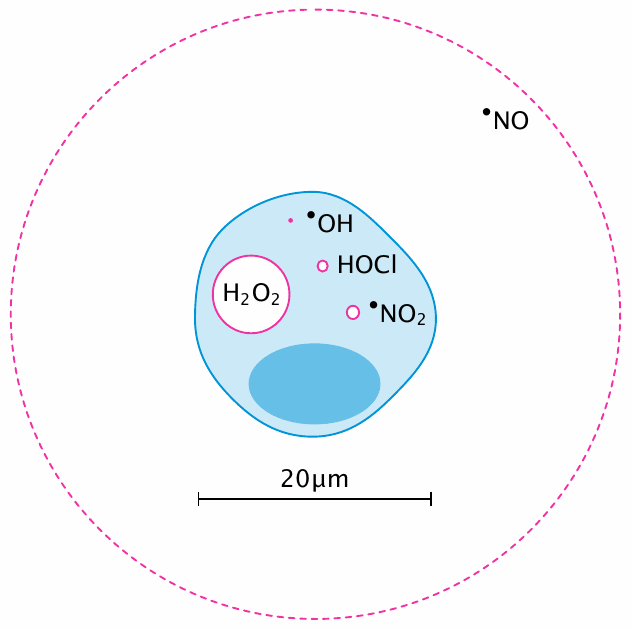
All animals are equal, but some are more equal than others—and so it is with reactive species: some are more reactive than others. Diffusion distances are a good way to illustrate this point: the greater a molecule’s reactivity, the shorter the distance it will travel between its creation and its consumption by chemical reaction. Thus, among the species shown in this slide, •OH is the most, and •NO the least reactive.108
The values shown are approximations; for example, H2O2 has a very much larger diffusion distance outside the cell—according to [149], 1.5 mm—than inside, where it is rapidly scavenged by peroxiredoxins (see next slide). The diffusion distance of •NO decreases at higher oxygen levels, but it is always large enough to encompass more than a single cell, which enables it to migrate between cells and thus transmit signals from one cell to another—which is, after all, one of its key functions (see section 9.3.6).
| 18.2.8 |
Example bimolecular reaction rate constants |
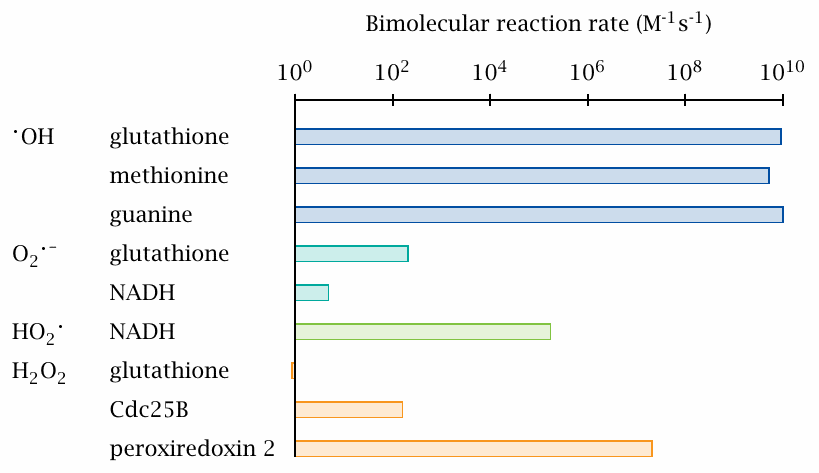
While the diffusion distance of a given RS reflects its tendency to react with anything at all that it may encounter in its path, a bimolecular reaction constant describes the reactivity when both of the reactants are specified. We can see that •OH reacts rapidly and indiscriminately with any of the offered reactants, each of which contains different functional groups.
O2•− is far less reactive; it is notable, however, that its protonated form (HO2•) reacts more readily with NADH. Since HO2• is also more membrane-permeant than O2•− and moreover has a higher redox potential (section 18.2.10), it also can react more readily with unsaturated fatty acyl residues and thus significantly contribute to lipid peroxidation (section 18.5).
Considering the prominent role of glutathione as a scavenger of reactive species, its low reactivity towards H2O2 may be surprising. Protein thiols such as those within the phosphatase enzyme Cdc25B may react much faster, and they are thus not effectively protected from H2O2 by glutathione alone. To ensure such protection, Nature invented the peroxiredoxins (see slide 18.7.4), which also employ thiol-disulfide chemistry yet have the highest reactivity toward H2O2 by far. They occur at high concentrations inside the cell, and at lower yet still relevant concentrations extracellularly; they have an important role in the body’s defense against reactive species.
The reaction rates in this slide were taken from [149] and [150].
| 18.2.9 |
The active site of the protein tyrosine phosphatase Cdc25B |
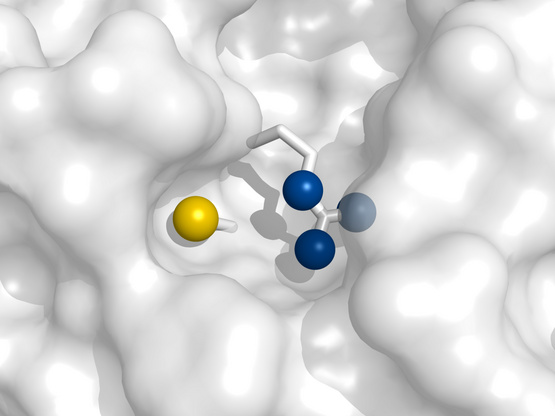
The preceding slide showed that the cysteine residue in the the active site of the protein tyrosine phosphatase Cdc25B is much more reactive toward H2O2 than is glutathione. As noted above, H2O2 reacts with the deprotonated (thiolate, R–S−) form of the cysteine side chain. The anionic thiolate is also part of the enzyme’s catalytic mechanism; it carries out the nucleophilic attack on the substrate’s phosphotyrosine residue. Next to the cysteine, we find an arginine residue, whose positive charge stabilizes the negatively charged thiolate and thus lowers its pKa to near physiological pH. In contrast, the pKa of the cysteine residue of glutathione is much higher, which explains its much lower reactivity toward H2O2.
While the high reactivity of Cdc25B toward H2O2 may seem accidental, it is plausible that it plays a role in the enzyme’s physiological regulation. The active site cysteine can be transiently inactivated by forming an intramolecular disulfide with another cysteine residue. Furthermore, Cdc25B plays a role in regulating cell division, and it is known that the redox state of the cell varies between different stages of the cell cycle. Generally speaking, there is considerable evidence implicating ROS in regulation and signaling. Many enzymes, ion channels, and receptors are modulated by them; and many cells express NADPH oxidase in order to generate ROS for signaling purposes [151].
While the reactivity of Cdc25B toward H2O2 has a straightforward explanation, the much greater reactivity of peroxiredoxins cannot be explained based on thiol group pKa alone but additionally involves specific mechanisms of transition state stabilization.
Structure rendered from 1qb0.pdb.
| 18.2.10 |
Standard redox potentials of selected radicals |
| Oxidised form | → | Reduced form | Δ E0′ (V) |
| •OH+H+ | +e− | H2O | 2.31 |
| R–O•+H+ | +e− | R–OH | 1.60 |
| HO–O•+H+ | +e− | H2O2 | 1.06 |
| R–O–O•+H+ | +e− | R–O–OH | 1.00 |
| R–S• | +e− | R–S− | 0.92 |
| H2C=CH–CH•–CH=CH2+H+ | +e− | H2C=CH–CH2–CH=CH2 | 0.60 |
| ascorbyl•−+H+ | +e− | ascorbate− | 0.28 |
| Fe+++ | +e− | Fe++ | 0.11 |
| dehydroascorbate | +e− | ascorbyl•− | −0.17 |
| O2 | +e− | O2•− | −0.33 |
| R–SS–R | +e− | R–SS•−–R | −1.50 |
| (water) | +e− | e− (solvated) | −2.87 |
The transfer of an electron to or from a radical is a redox reaction, so we can assign it a redox potential. This table—excerpted from a longer one in [152]—shows some representative examples.109 The redox potential of a reaction is proportional to its free energy; and as you know, the free energy alone does not tell us much about the reaction rate. However, the transfer of a single electron to or from a free radical is generally kinetically facile, so that most radical reactions that are energetically feasible can also be observed in practice.
As discussed in section 6.5, a positive redox potential corresponds to a negative free energy; therefore, in this table, •OH has the strongest ‘pull’ toward free electrons and thus will oxidize any other reduced species listed. On the other hand, free solvated electrons, which can be generated by ionizing radiation (see slide 18.2.1), are not bound to anything that could lower their free energy, and thus quite understandably can reduce any of the oxidized species.
Based on these considerations, we can predict that H2C=CH–CH2–CH=CH2 (1,4-pentadiene), which is a model compound for unsaturated fatty acids, can be oxidized to the carbon-centered radical (–CH•–) by the thiyl radical (R–S•) or any of the reactive species above it; and indeed the literature implicates all of these species in such reactions as part of lipid peroxidation (see section 18.5).
Another point to note is the position of superoxide relative to other radicals. Both of the ascorbate redox couples are above superoxide; therefore, ascorbate does not reduce oxygen to O2•−, which in light of the ubiquity of O2 would be a major calamity. The same holds for all other major antioxidants/radical scavengers. On the other hand, the disulfide radical anion (R–SS•−–R) is further down and therefore does reduce O2 to O2•−; this was already noted above in slide 18.2.3.
| 18.2.11 |
Some radicals are stabilized by resonance |
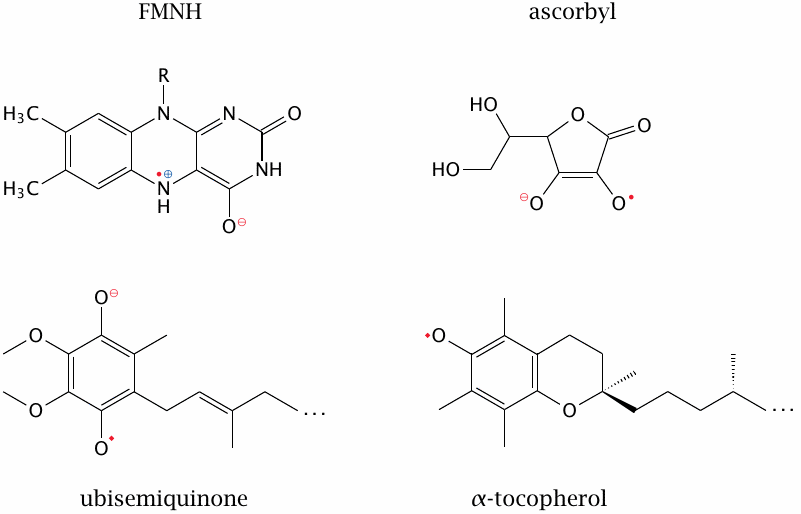
While the reactivity of radicals is generally high, it can be mitigated by conjugating the unpaired electron to a system of delocalized π-electrons. All of the four examples shown here occur in human metabolism, and three of them are in fact produced in reactions that scavenge other, more reactive radicals; these relatively stable radicals can then be disposed of in an orderly manner that avoids damage to random bystander molecules. This will be discussed in more detail in section 18.7.
While FMNH is not usually listed among the radical scavengers, the stable containment of its unpaired electron is important for its biological function nevertheless, since it facilitates the transfer of single electrons in the respiratory chain. Ubisemiquinone functions both as a radical scavenger and as a redox cofactor within the respiratory chain. Both carriers do leak a certain fraction of the transferred electrons to molecular oxygen, however, and the mitochondria must deal with this oxydative stress (see slide 18.3.4).
| 18.3 |
Major sources of reactive species in vivo |
While multiple enzymatic and non-enzymatic reactions produce reactive species in vivo, two sources that generate them in particularly large quantities are phagocytes (macrophages and granulocytes) as well as, more ubiquitously, the respiratory chain.
| 18.3.1 |
NADPH oxidase initiates ROS formation in phagocytes |
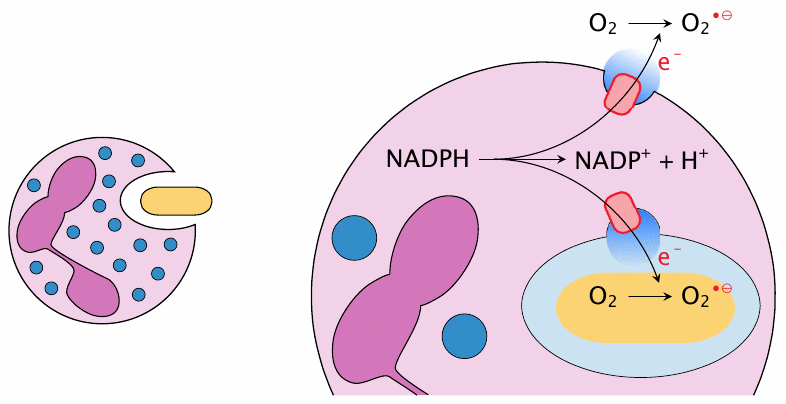
The most striking example of RS generation on purpose are phagocytes (both macrophages and granulocytes). These cells ingest pathogens and then destroy them using a mixture of aggressive chemicals, of which reactive oxygen and nitrogen species are an important element.110 To this end, the phagosome that contains the microbe fuses with specialized vesicles (granules) that harbor NADPH oxidase, a membrane protein that abstracts electrons from cytosolic NADPH and makes them available for the reduction of O2 to O2•− inside the phagosome [153]. The activation of NADPH oxidase causes a sudden increase in O2 consumption that is known as the respiratory burst. Additional ROS are then generated from O2•− (see next slide).
NADPH oxidase can also trigger the extracellular production of ROS. This occurs during degranulation and is important in the defense against infectious pathogens that are too large for ingestion, for example fungi like Aspergillus flavus. During both intra- and extracellular activation of NADPH oxidase, the transmembrane electron flow must be balanced by the opening of cation channels for charge compensation; otherwise, the ensuing membrane potential would soon put a stop to further electron generation.111
| 18.3.2 |
O2•− gives rise to other reactive oxygen species |
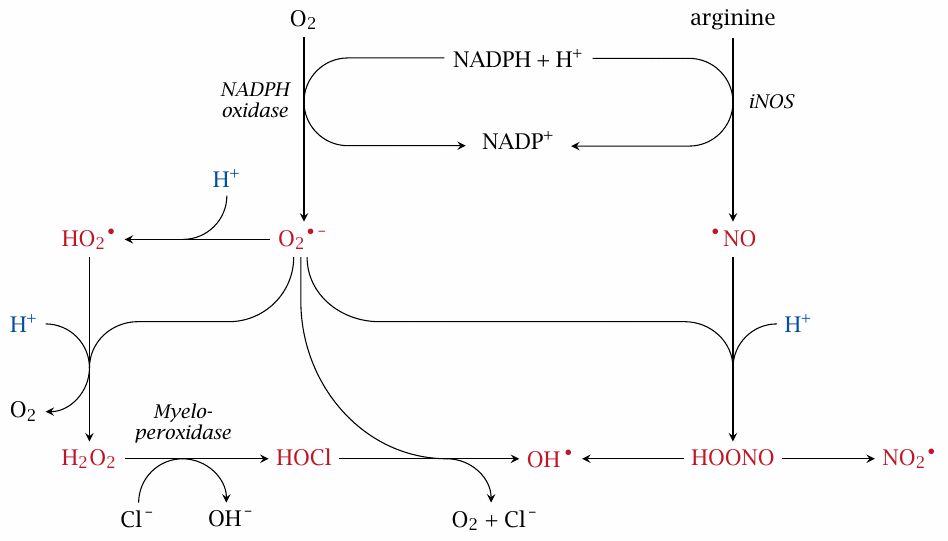
NADPH oxidase gets phagocyte ROS generation started, but there is more. A second ‘starter’ radical is nitric oxide (•NO), which is produced by inducible nitric oxide synthase (iNOS) in the cytosol but can easily diffuse across membrane barriers. O2•− and •NO can combine into peroxynitrite (HOONO), which in turn can decay to •OH and nitrogen dioxide (•NO2), both of which are highly reactive. A second pathway of •OH generation is via H2O2 and HOCl, which is supplied by myeloperoxidase.
While all of the molecules shown here in red are considered reactive species, O2•− and •NO are less reactive than the others, which likely are the main antimicrobial effectors. Note how the acidic pH inside the phagosomes comes into play:112 protonation of O2•− produces HO2•, a more reactive and more membrane-permeant radical that is also the precursor for H2O2 and HOCl. Likewise, peroxynitrite becomes more reactive and membrane-permeant through protonation.
From this scheme, it is apparent that disruption of NADPH oxidase activity would significantly disrupt ROS formation, and indeed gene defects for this enzyme impair immune defense, particularly against bacterial pathogens.113 Interestingly, a genetic deficiency of myeloperoxidase has no reported phenotype, suggesting that HOCl as such is not a crucial antimicrobial effector.
| 18.3.3 |
Lessons from ROS generation in phagocytes |
- ROS are produced in large amounts for killing microbes, even though they will also damage host cells
- ROS generation starts with reducing power, and often (as in this case) with enzymatic reactions
- Once primary RS have been generated—here, O2•− and •NO—they tend to spontaneously generate secondary ones
- pH matters—the weakly acidic endosomal pH seems optimized for generating peroxynitrite and HO2•
While ROS are an effective means for killing microbes, they also can cause substantial damage to the host, particularly in chronic inflammation. What is more, inflammation will generate ROS even when no pathogen is present; this occurs in autoimmune diseases like rheumatism, as well as in inflammation caused by crystalline deposits of uric acid (gout), silicates (miner’s lung), or asbestos (asbestosis). While long-running gout manifests itself mainly in the destruction of the afflicted joints, other chronic inflammatory diseases may give rise to tumors, most likely through the genetic damage inflicted by ROS [154].114
Since phagocytes generate RS on purpose, this picture does not show any mechanisms for scavenging them. In contrast, most other cells have mechanisms for scooping up the RS before they can do too much harm. However, when these radical scavengers become depleted by toxic xenobiotics or drug metabolites, cell and organ damage will result. In this context, it is interesting to note that activated phagocytes—granulocytes, in particular—tend to be sacrificed in a single round of activation; the large amount of ROS produced in the full-scale respiratory burst will seal the fate of both the microbe and the granulocyte.115
| 18.3.4 |
Production of reactive oxygen species in mitochondrial respiration |
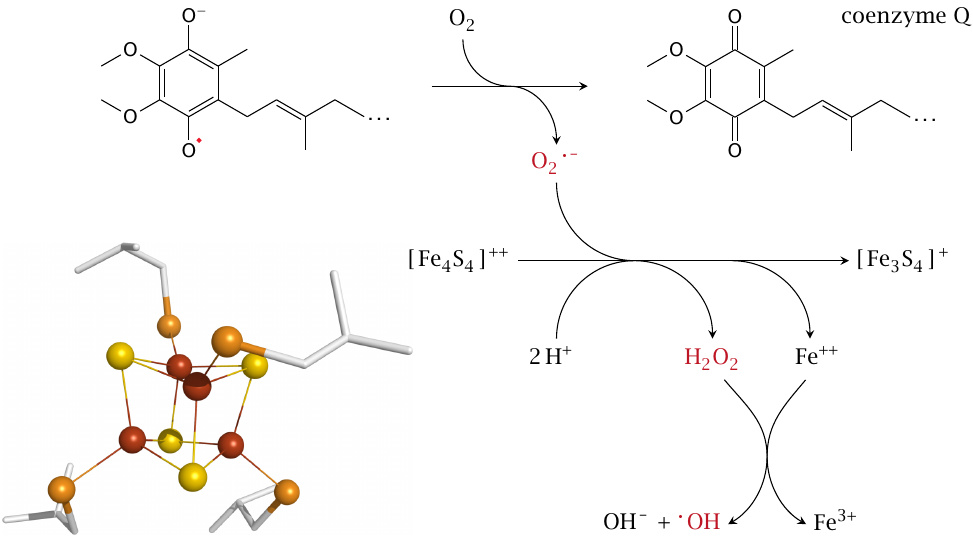
In the respiratory chain, ROS generation occurs as a side effect rather than on purpose. The diffusible carrier coenzyme Q can lose an electron to molecular oxygen [156], as can FMN (see section 6.4.3). The superoxide produced can react with FeS clusters, which occur in great numbers in the mitochondrial respiratory chain (see section 6.4). This reaction reduces O2•− to H2O2 and releases Fe++ from the cluster. As discussed in slide 18.2.5, Fe++ can then react with H2O2 to produce an •OH radical [157]. Alternatively, Fe++ may react with alkyl peroxides (R-OOH) to produce alkoxyl radicals (R–O•, slide 18.2.6), which may contribute to lipid peroxidation.
The pathway outlined here illustrates how, in a few short steps, a relatively benign reactive species (O2•−) may turn into something very harmful. Normally, most of the superoxide produced in the respiratory chain does not end up liberating iron but instead is scavenged, as shown in slide 18.7.6. However, when the cell’s antioxidant defenses are depleted, the process depicted here will indeed occur and damage the cell.
| 18.3.5 |
Mitochondrial energy state and ROS formation |
- When ATP consumption is low, proton and electron transport chain back up
- Backed-up electrons will leak and produce more O2•−
- O2•− activates uncoupling proteins, which will lower the proton-motive force and the ATP yield, but increase electron transport
- Increased O2•− formation has been observed in pancreatic β-cells in type 2 diabetes
As discussed in Chapter 6, mitochondrial electron transport and proton transport are intimately linked, and one cannot proceed without the other. If extruded protons are not used toward ATP synthesis, the proton gradient will rise until the electron transport chain (ETC) can no longer pump protons against it. This will cause the electrons in the ETC to back up also and be leaked to O2, producing O2•−.
The activation of uncoupling proteins (UCPs) by O2•− will dissipate the proton gradient, restore electron transport, and reduce electron leakage. UCPs thus provide a safety valve that guards against excessive O2•− formation. As a side effect, however, UCP activation will also decrease the yield of ATP produced through the oxidation of a given quantity of substrate.
As we had seen in slide 13.2.8, the utilization of glucose toward ATP synthesis is required to trigger insulin secretion from pancreatic β-cells. If for some reason the generation of O2•− in β-cells should be increased, and thus the ATP yield lowered by UCP activation, this could interfere with insulin secretion. Some experimental evidence suggests that such an effect occurs in type 2 diabetes [158], but the causes of increased O2•− formation are not well understood.116
| 18.4 |
Radical reactions with nucleic acids and proteins |
Considering the large number of reactive species and the great variety of macromolecules, the possible reactions between them are of course innumerable; we will confine the discussion here to some instructive and biologically relevant examples.
| 18.4.1 |
Hydroxyl radicals can modify DNA bases |
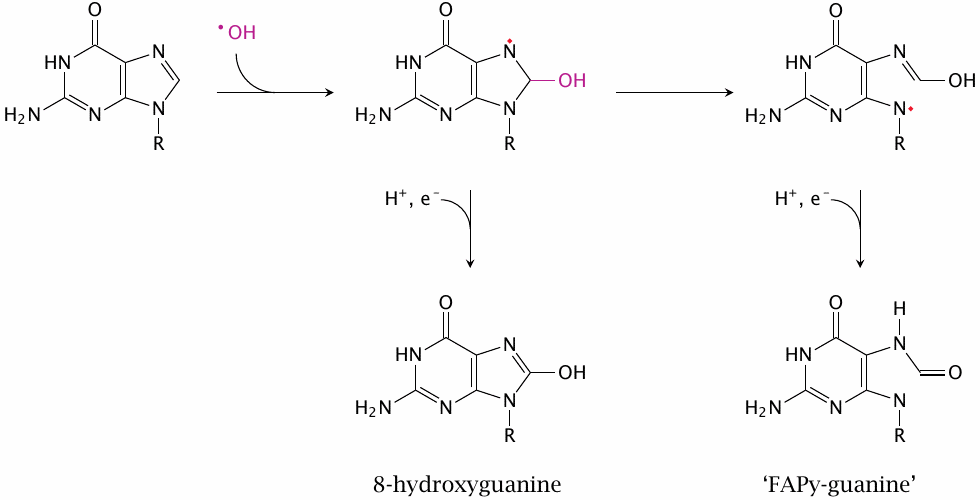
While •OH radicals can indeed react with any biomolecule, the most consequential reactions will be those with DNA, due to its unique biological function: while every other biomolecule can be replaced when damaged, DNA cannot. The reaction shown here involves the N7 of guanine; other positions in this base can react as well, as can the other bases found in DNA. Such base modifications may result in mispairing during DNA replication, which will then give rise to mutations.
The prefix ‘FAPy’ is shorthand for 2,6-diamino-4-hydroxy-5-formamidopyrimidine; while the name ‘FAPy-guanine’ is not very precise, it is commonly used in the literature.
| 18.4.2 |
Hydroxyl radicals can break DNA strands |
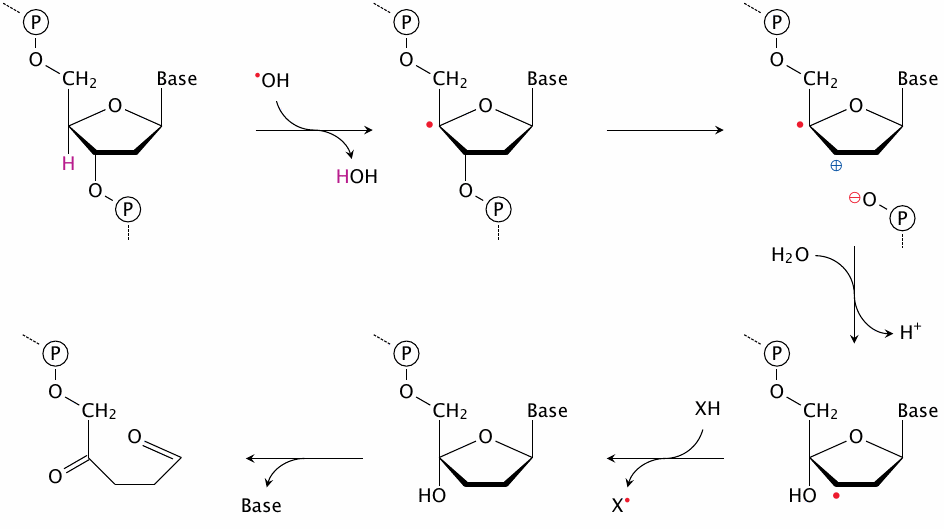
This slide, which is based on a scheme in [159], shows how the reaction between a deoxyribose moiety and •OH can lead to cleavage of the DNA strand. The cleavage event (shown at the top right) may be followed by reactions other than the ones shown here, but this variation would not seem particularly relevant with respect to the overall biological effect.
While single-strand breaks are in most cases successfully repaired, double-strand breaks present the cell with a much harder problem, and there is a higher chance that repair fails or results in mutations, including chromosome breaks and translocations. The likelihood that one single-strand break will be located close to a second one on the opposite strand—or in other words, that it be part of a double-strand break—will increase with the local concentration of •OH; thus, genetic damage due to ionizing radiation increases stronger than linearly with dosage.117
| 18.4.3 |
Protein modification by reactive oxygen species |
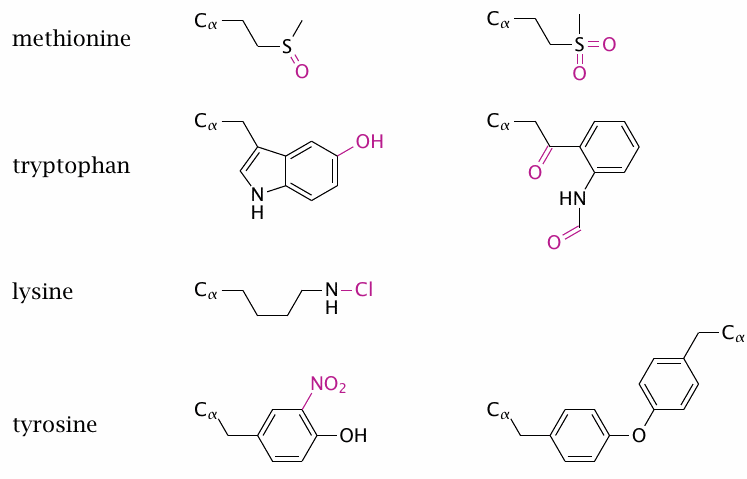
Most cells contain much more protein than nucleic acids, and for this simple reason protein will react with a greater share of reactive species. All amino acid side chains in proteins may be modified, but some make particularly good targets. Examples are methionine, which can react with H2O2 or other ROS to produce methionine sulfoxide (top left) or the sulfone (top right). The former, but not the latter, can be reduced by peptide methionine sulfoxide reductase, which is itself reduced by thioredoxin. However, most other protein modifications are irreversible and likely to result in the degradation of the entire protein molecule.
The aromatic amino acids are susceptible to ring halogenation, which is mediated by HOCl or HOBr, nitration, which is caused by •NO2, and also to hydroxylation by •OH. With tryptophan, oxidative ring cleavage can give rise to N-formylkynurenine (second from the top, right). Tyrosine may form dimers (bottom right).118 Nitrogen atoms in amino acid side chains may react with HOCl to generate chloramines, shown here using lysine as an example. Chloramines are unstable and will in turn react as oxidizing agents.
Another preferred target of oxidative modification are cysteine residues; these react according to the general schemes shown earlier for thiol groups (see slides 18.2.3 and 18.2.4). Many other side chain modifications are possible, as are modification and even cleavage of the protein backbone. While in most cases the effects are non-specific, some proteins can be decisively activated or inhibited by these oxidative modifications; for example, the protease inhibitor α1-antitrypsin is itself strongly inhibited by peroxynitrite [160].
In addition to the modifications shown here, proteins can also give rise to carbon-centered radicals (R–CH•–R), for example by reaction with •OH radicals. These may either combine with radicals (particularly O2), or they may be repaired by antioxidants. It is interesting to note that both ascorbic acid [161] and urate [162,163] can react with radicals of aromatic acid side chains in proteins much faster than can glutathione, and fast enough to compete with O2 radical recombination. This may be a key function of these two antioxidants.
| 18.5 |
Lipid peroxidation |
Like proteins and nucleic acids, lipids may also be modified by reactive species. Most lipid molecules reside in cell membranes, lipid droplets, and lipoproteins; they are therefore most susceptible to reactive species that are hydrophobic enough to partition into these environments. Similarly, protection of lipids from degradation by such reactive species requires lipophilic radical scavengers, in particular α-tocopherol (vitamin E) and ubiquinone. Once expended, these lipophilic scavengers can be regenerated by hydrophilic reducing agents at the membrane interface (see slide 18.7.13). Accordingly, when hydrophilic reductants are depleted, for example by redox cycling (see slide 9.4.2),lipophilic scavengers will soon be as well.
Lipid-rich compartments may also host hydrophobic complexes of transition metals. In particular, free heme can associate with lipid membranes and lipoproteins, and within these it can promote the formation of reactive species. Both depletion of scavengers and accumulation of transition metals will set the stage for lipid peroxidation.
| 18.5.1 |
Self-sustained lipid peroxidation induced by peroxyl radicals |
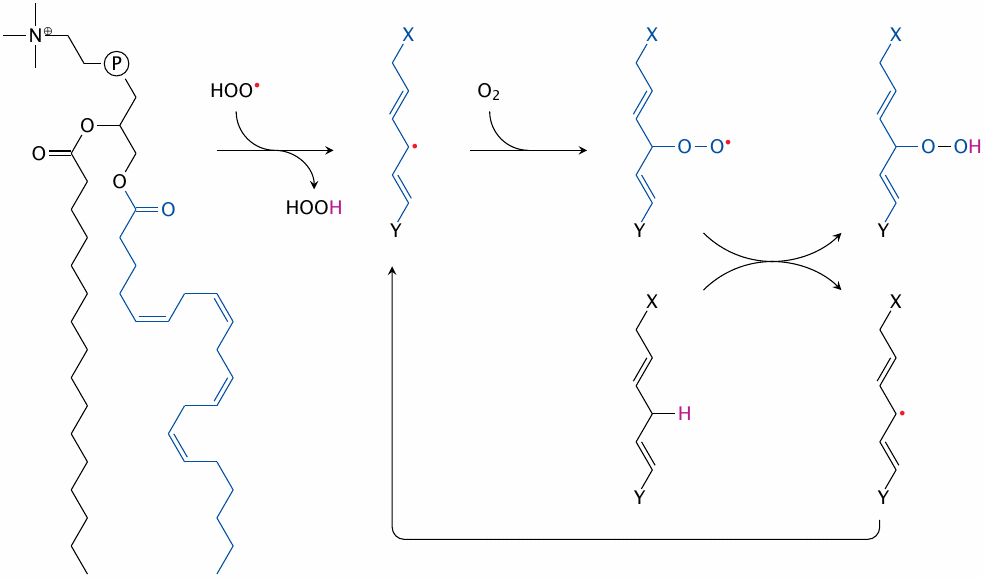
Lipid peroxidation begins with the conversion of a fatty acyl methylene group to a carbon-centered radical. Particularly susceptible are bisallylic methylene groups in multiply unsaturated fatty acyl residues that are wedged between two adjacent double bonds.119 The reduction potential of bisallylic methylene groups is lower than that of hydroperoxyl (HOO•), alkoxyl (RO•) or alkylperoxyl (ROO•) radicals (see table in slide 18.2.10); any of these can therefore steal an electron (with its accompanying proton) from a bisallylic carbon.
The carbon-centered radical thus created may then combine with O2, which produces an alkylperoxyl radical that can in turn react with another bisallylic methylene group to regenerate a carbon-centered radical. This creates a reaction cycle that converts unsaturated fatty acids to lipid peroxides, and which needs only molecular oxygen to sustain itself. O2 is always readily available; it is indeed more soluble in lipid membranes or droplets than in water. Therefore, in lipid peroxidation, a single radical that goes unchecked can trigger the degradation of a large number of lipid molecules and thereby disrupt membrane stability.
| 18.5.2 |
Toxic products of lipid peroxidation: hydroxynonenal |
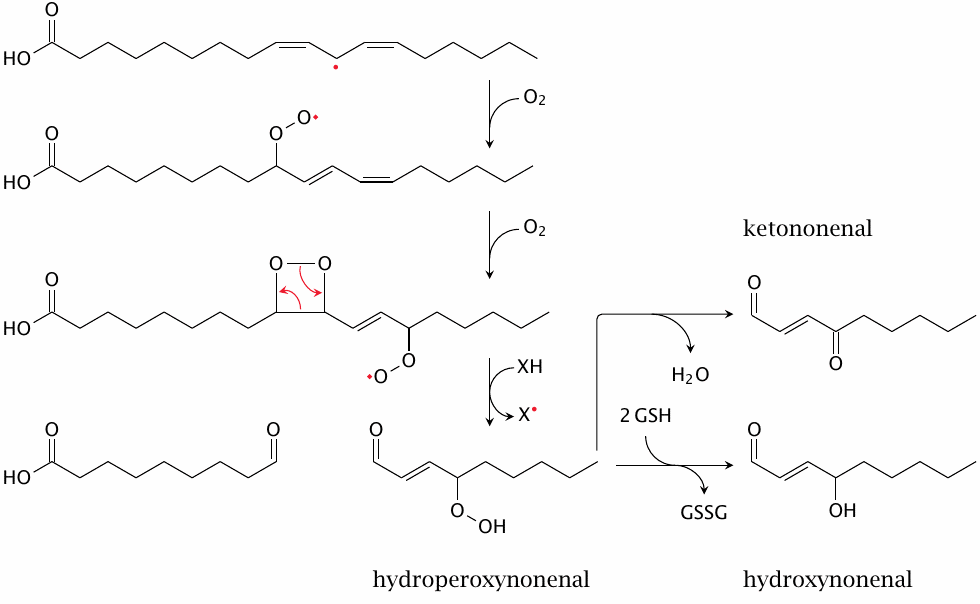
In addition to simply converting unsaturated fatty acyl residues to hydroperoxides, as shown before, lipid peroxidation can also give rise to toxic breakdown products such as hydroxynonenal. The reaction pathway shown here is an example; other pathways to this product or to similar ones are possible [164]. The reactions up to the hydroperoxyl derivative are spontaneous; its reduction to the hydroxyl may be catalyzed by glutathione peroxidase (see slide 18.7.7). While all three of the nonenal derivatives shown are toxic and reactive, hydroxynonenal seems to be most prominent [165].
Also note, both here and in slide 18.5.4, how lipid peroxidation may induce the formation of conjugated double bonds; these can be detected e.g. by UV absorption (see slide 18.6.4).
| 18.5.3 |
Hydroxynonenal cytotoxicity in cell culture |
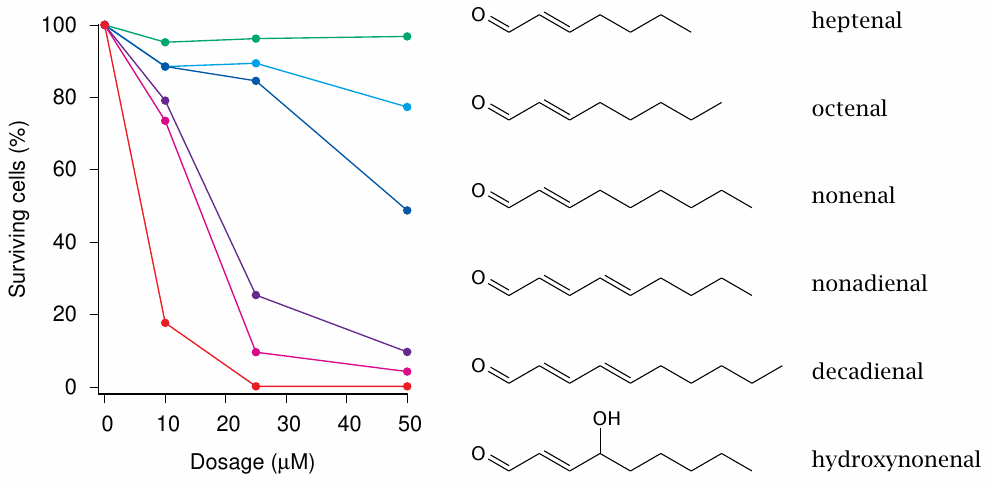
The experiments shown in this slide [166] show that among a group of structurally similar compounds hydroxynonenal is the most toxic. A recent study [167] shows that hydroxynonenal can activate receptor-medicated apoptosis and also gives an overview of other possible mechanisms of toxicity.
| 18.5.4 |
Toxic products of lipid peroxidation: malondialdehyde |
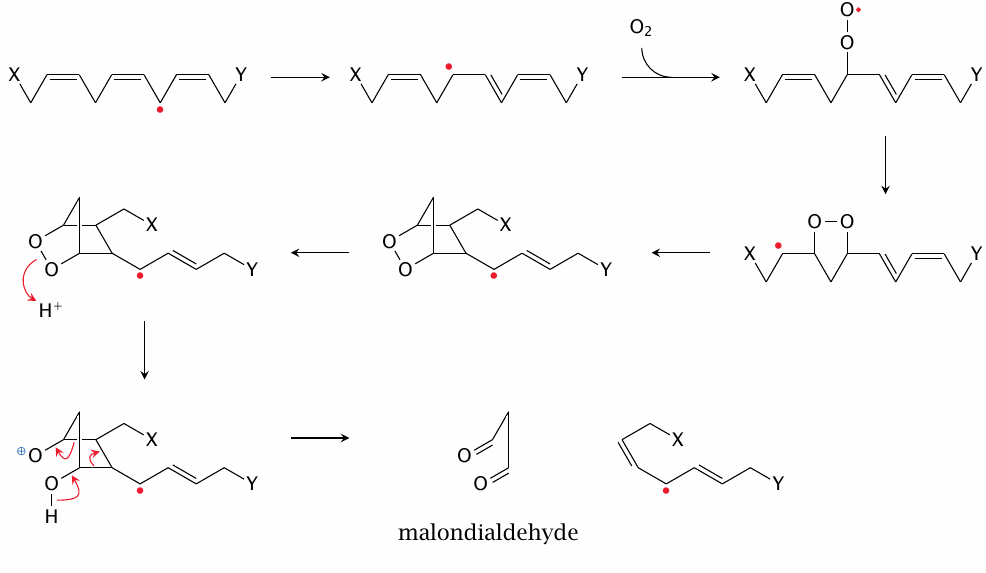
Aside from hydroxynonenal, another prominent fragmentation product of lipid peroxidation is malondialdehyde. The pathway depicted here is based on [168], with several details filled in by me in a somewhat speculative manner. In particular, it seems possible that the second radical—the one that I drag along all the way to the second final product in this scheme—also reacts earlier on by combining with O2 or in some other way.
| 18.5.5 |
Formation of nucleobase adducts by hydroxynonenal and malondialdehyde |
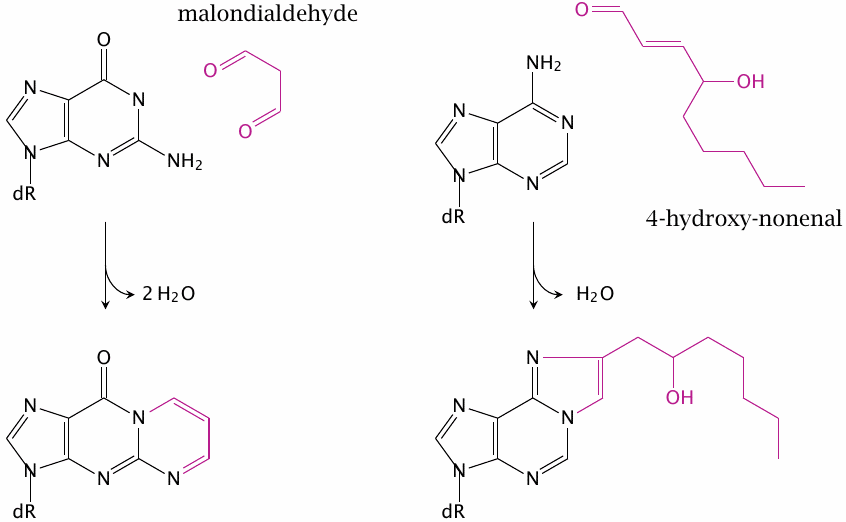
Both of the lipid peroxidation products shown above can form adducts with DNA bases. Shown here is the reaction of malondialdehyde with guanine and that of 4-hydroxynonenal with adenine.
Structures drawn after [154,169] and intended as examples only; similar adducts may form from other bases and lipid peroxidation products. DNA adducts like these may cause mutations; this is one of the mechanisms by which chronic inflammation, with its attendant release of reactive oxygen species from phagocytes, may promote cancer [154]. This disease mechanism is particularly common in asbestosis, which is caused by the inhalation of fine, needle-shaped asbestos crystals. These crystals cause a chronic inflammation in the tissues within and surrounding the lungs. As generations of macrophages ingest and try in vain to destroy these crystals, the concomitant formation of ROS often induces either bronchial carcinoma or pleura mesothelioma.
| 18.5.6 |
Detection of malondialdehyde with thiobarbituric acid |
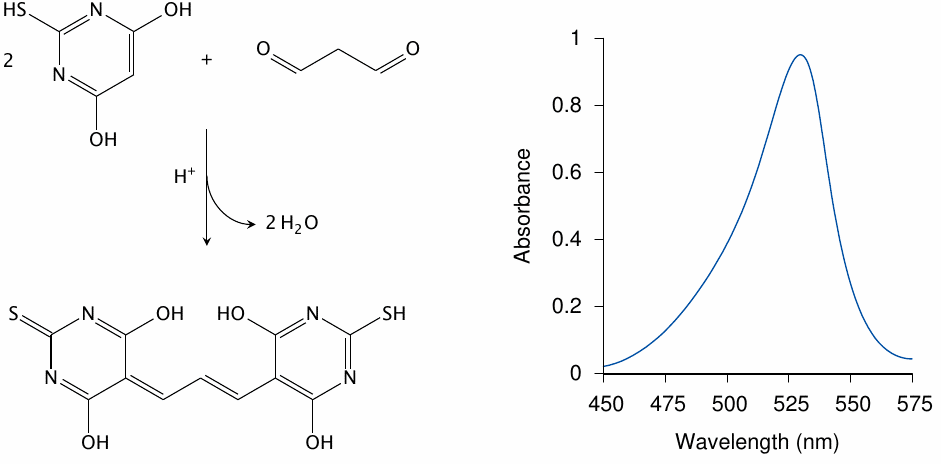
Apart from its role in pathogenesis, malondialdehyde is also relevant as a diagnostic marker of lipid peroxidation, since it forms a chromophore when reacted with thiobarbituric acid. This test has been widely used in experimental studies on lipid peroxidation (see for example [170–172]).
It has been pointed out that the reaction conditions of the thiobarbituric acid test, which involve heat and acid, will cause more malondialdehyde to form than was present in vivo or under biologically relevant in vitro conditions. However, the oxidized precursors for the additional malondialdehyde released during the reaction with thiobarbituric acid will still have to be generated by lipid peroxidation beforehand, so that the test remains a useful measure of lipid peroxidation [173].
Reaction scheme drawn after [174], and spectrum after [175].
| 18.5.7 |
Formation of inflammatory mediators by enzymatic lipid peroxidation |
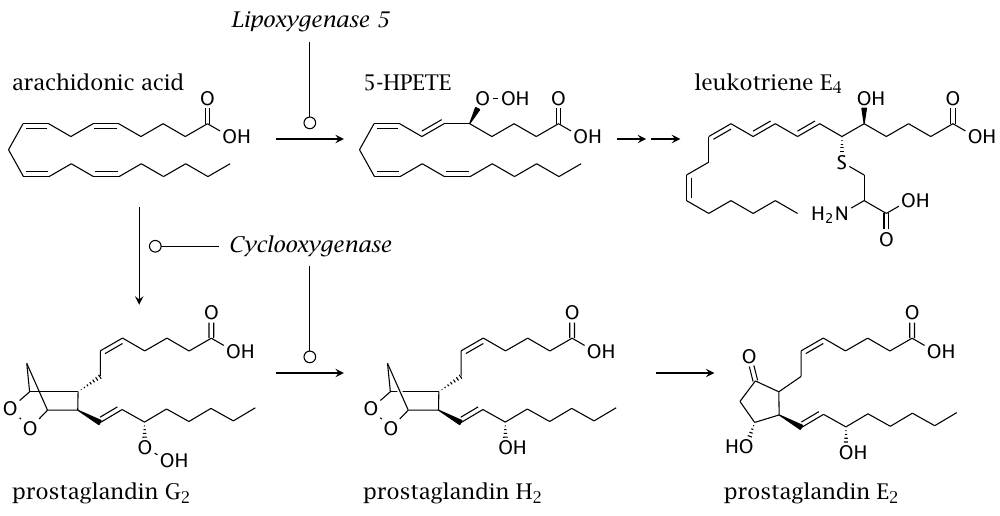
Considering the harmful effects of runaway lipid peroxidation, it stands to reason that life should have evolved sensing mechanisms to detect its products. In an interesting twist, Nature then evolved enzymes that generated these products on purpose as signaling mediators, of which this slide shows some examples. The formation of prostaglandins is initiated by cyclooxygenase, whereas leukotrienes are formed downstream of lipoxygenases. Both pathways begin when arachidonic acid is released from membrane-associated phospholipids by cytosolic phospholipase A2, which is itself activated by calcium signals.
Prostaglandins and leukotrienes are important mediators of inflammation, acting mostly through G protein-coupled receptors. Inhibitors of the enzymes that produce them are widely used in medicine; in particular, cyclooxygenase inhibitors include popular drugs such as acetylsalicylic acid (Aspirin) and acetaminophen (Tylenol).120
You may have noted the cysteine residue in the structure of leukotriene E4. This is introduced as part of glutathione, whose glycine and glutamate residues are then cleaved. Glutathione conjugation is a common detoxification pathway (chapter 19); thus, it appears that evolution coopted not only the lipid peroxidation products themselves, but also their detoxification products as signaling molecules.
| 18.5.8 |
Lipoxygenases use iron to abstract H• from the substrate |
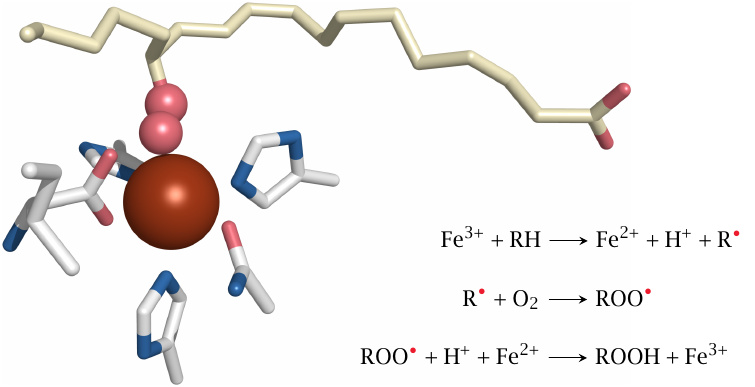
Lipoxygenases employ a transition metal (iron) to facilitate lipid peroxidation. The iron ion, whose ligands within the active site include three histidine residues and the C-terminal carboxyl group of the protein, alternates between the ferrous (Fe++) and ferric (Fe+++) oxidation states. In the structure shown here,121 the carbon-centered radical introduced into the fatty acid substrate in the first step has already combined with O2; since the hydrogen atoms are missing from the structure, we can’t know if the third step of the reaction has already been completed.
It was noted earlier that transition metals can also facilitate non-enzymatic lipid peroxidation though generating hydroxyl or alkoxyl radicals by Fenton chemistry. If I understand the literature on lipoxygenase catalysis correctly, then in this case it is the iron ion itself that attacks the bisallylic methylene group. For this to work, the enzyme would have to somehow raise the redox potential of the iron to above that of the bisallylic carbon. In this context, it is noteworthy that some iron complexes, e.g. iron phenanthroline [152], indeed have such a high redox potential, and thus might act similarly to lipoxygenase. I do not know whether this also applies to any non-enzyme iron complexes that are likely to occur in vivo.
| 18.5.9 |
A tyrosyl radical initiates the cyclooxygenase reaction |
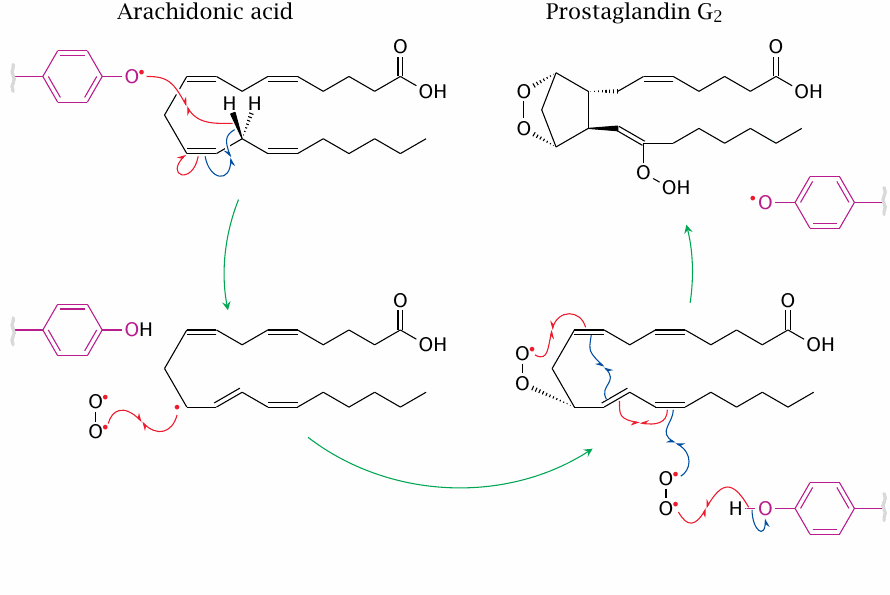
Like transition metals, radicals also figure in both non-enzymatic and enzymatic lipid peroxidation. In the first reaction catalyzed by cyclooxygenase, the substrate is converted to a carbon-centered radical by a tyrosine phenoxyl radical in the active site. The latter is regenerated after the introduction of two oxygen molecules into the substrate.
The second reaction catalyzed by cyclooxygenase—the reduction of the hydroperoxide formed in the first one to a hydroxide, cf. slide 18.5.7—occurs in a separate active site and requires glutathione; it resembles the reduction of lipid peroxides formed by glutathione peroxidase.
| 18.6 |
Singlet oxygen |
The term singlet oxygen refers to two different excited states of molecular oxygen (O2), which can arise in vivo by either photoactivation [176] or through enzymatic reactions, for example in phagocytes [177]. Of the two mechanisms, photoactivation, particularly by porphyrin molecules in diseases such as porphyria cutanea tarda (see section 17.3.3), is the more clinically important.
| 18.6.1 |
Photoactivated generation of singlet oxygen by porphyrins |
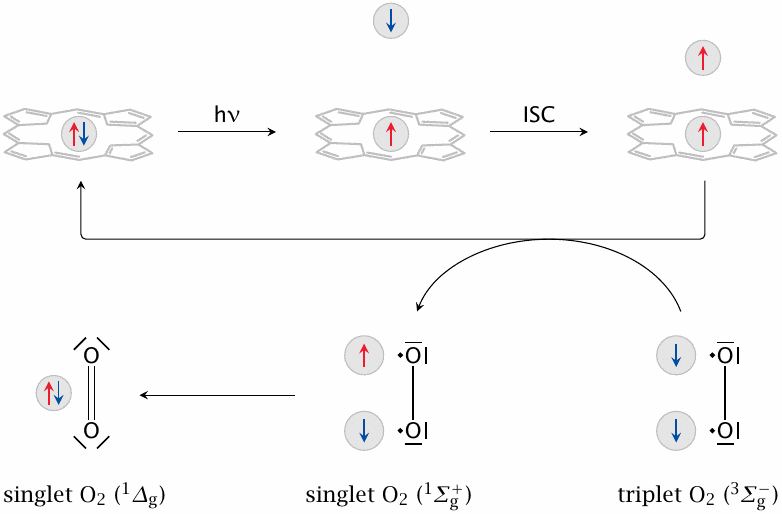
Free porphyrins (but not metal-containing ones such as heme) are effective mediators of singlet oxygen generation. This slide summarizes the basic photophysics of this process. Balls represent electrons, and arrows the parallel or antiparallel orientations of their spins.
The ground state of a porphyrin molecule is a singlet state, which means that each electron is matched by another one that has an antiparallel spin. When the molecule absorbs a photon (hν), one of its electrons is promoted into a different orbital of higher energy, where it may undergo spin inversion (inter-system crossing, ISC) by interacting with other electron orbitals. This results in a triplet state, which has two unpaired electrons with parallel spins.
The spin-inverted electron is now stuck in its own high-energy orbital, since the Pauli principle prevents it from joining another electron with the same spin. However, the porphyrin molecule can revert to the ground state by trading spins with molecular oxygen (O2), whose lowest energy state is also a triplet state. When this occurs, both molecules transition to their respective singlet states. While the porphyrin will lose some excitation energy in this interaction, the oxygen will actually gain energy; a part of that energy will be released again when the two unpaired but now spin-parallel electrons join the same orbital. This second form of singlet oxygen (1Δg) is therefore more stable than the first one and is the predominant species in vivo.
The Lewis structures shown here to represent the various forms of O2 are necessarily simplifications. While they do capture the difference in the pairing of π-electrons between triplet and singlet oxygen, in all cases the electrons sit in orbitals that are shared between both atoms but anti-bonding. Also note that both singlet states are higher in energy than the triplet (ground-state) form [176,178].
The following slides will provide some mechanistic detail on the reactions of singlet oxygen that may lead to cell and tissue damage. Here, we just note that the photosensitized formation of singlet oxygen by porphyrins lies at the heart of the skin damage that occurs in porphyria cutanea tarda and other enzyme defects in the heme synthesis pathway [179].122
| 18.6.2 |
Singlet oxygen reacts readily with non-radicals |
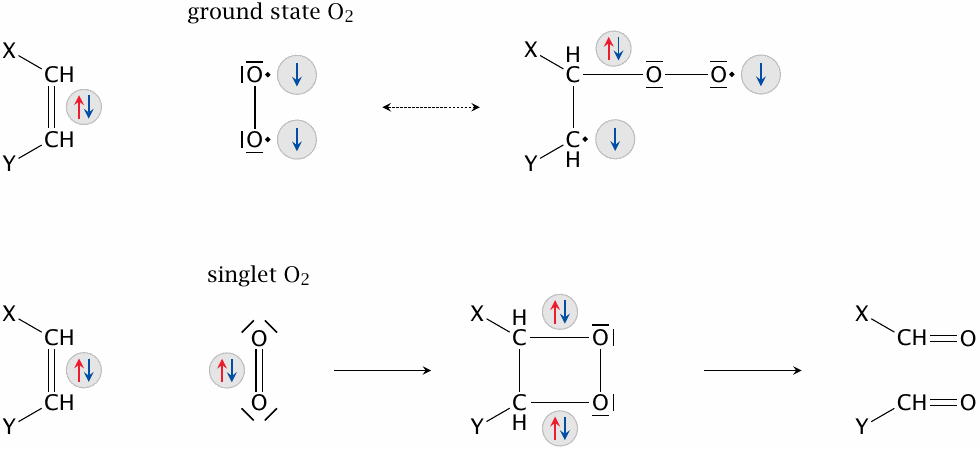
Ground-state (triplet) oxygen can’t productively engage electron pairs with opposite spins—two electrons will always remain unpaired and prevent the formation of a low-energy reaction product. This is a good thing—otherwise, our hair, our pants, and indeed the world around us would be remarkably prone to spontaneous combustion. Singlet oxygen, in contrast, can engage paired electrons; in addition, the energy that it contains in excess of ground-state oxygen can be used toward activating such a reaction. It may, as shown here, insert itself into a double bond with subsequent cleavage, react similarly across conjugated double bonds, or simply insert itself into a single C–H bond to produce a hydroperoxide (see next slide). Such reactions may involve nucleotides, amino acids, and lipids.
| 18.6.3 |
Singlet oxygen and transition metals in photoactivated lipid peroxidation |
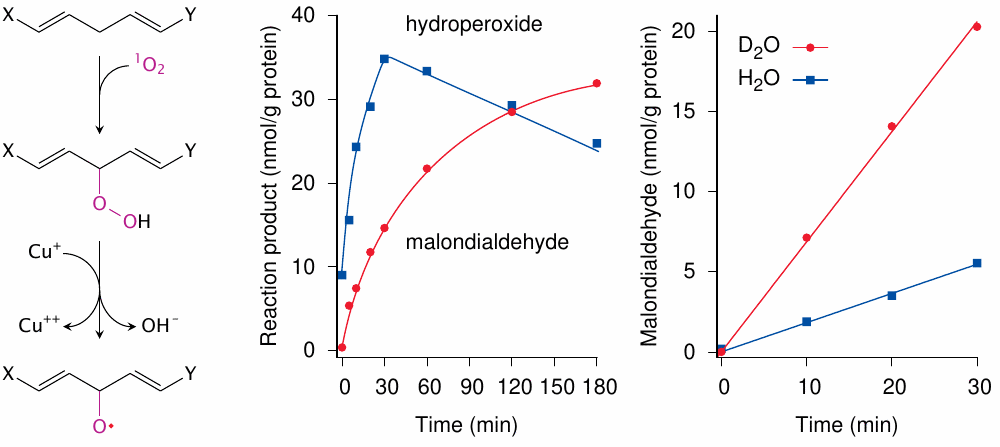
In the experimental study shown here [181], red blood cell membranes were UV-irradiated in the presence of a semisynthetic porphyrin (hematoporphyrin) and copper ions. Left: Photosensitization produces singlet oxygen (1O2), which directly reacts with an unsaturated fatty acid to form a hydroperoxide. The latter is activated to an alkoxyl radical by Cu+,123 and a lipid peroxidation cycle starts. Center: Lipid hydroperoxides initially accumulate but then decay, which produces malondialdehyde (see slide 18.5.4). Right: singlet oxygen is readily quenched by water, but less so by heavy water (D2O). The greater yield of malondialdehyde in the presence of heavy water supports the role of singlet oxygen in the overall process.
Apart from confirming the role of singlet oxygen in lipid peroxidation, the heavy water experiment also illustrates that singlet oxygen is not very effective in an aqueous environment; it is more long-lived and thus more effective in a hydrophobic one. While porphyrins vary in hydrophobicity, the more hydrophobic ones tend to cause more damage, probably because they generate singlet oxygen within cell membranes. In this context, it is noteworthy that vitamin A, which is hydrophobic and contains multiple double bonds that may react with and thus scavenge singlet oxygen, can substantially mitigate skin damage in porphyria; this is used in the clinical treatment of such patients [179].
| 18.6.4 |
UV-induced lipid peroxidation and membrane damage in erythropoietic protoporphyria |
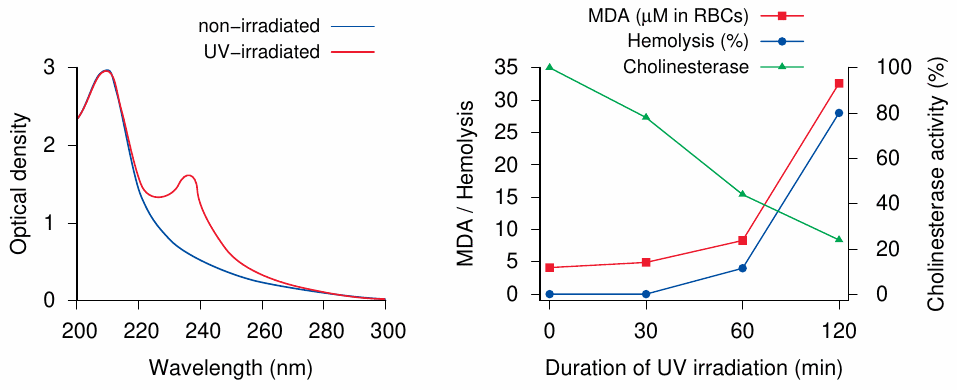
The experiments shown here [182] are similar to the ones in the preceding slide. Erythrocytes were taken from patients with erythropoietic protoporphyria, which is caused by a deficient ferrochelatase enzyme (see section 17.3.5), and irradiated with UV light. Left: After irradiation, the red blood cell membranes displayed a new absorption peak, which corresponds to conjugated double bonds formed during lipid peroxidation (cf. slide 18.5.2 and 18.5.4). Right: malondialdehyde formation, which indicates breakdown of membrane lipids, parallels hemolysis (that is, red blood cell lysis). The activity of cholinesterase, a representative membrane protein, drops off concomitantly with irradiation-induced lipid peroxidation.
The use of red blood cells in this experiment was likely based on experimental convenience more than on relevance for disease; as with other porphyrias, the skin is the major target organ. However, protoporphyrin—the porphyrin which accumulates when ferrochelatase is missing—does bind to plasma proteins and lipoproteins, which would tend to retain it in the circulation, and it has been said that indeed capillary lesions may be the primary event at least in this particular porphyria [183].
| 18.7 |
Protective mechanisms and molecules |
- Metal sequestration (Fe, Cu)
-
Enzymes
- Superoxide dismutase
- Catalase
- Glutathione peroxidase family
- Peroxiredoxins, glutaredoxins, thioredoxins
-
Small molecules
- Endogenous: glutathione, uric acid, bilirubin, coenzyme Q
- Exogenous: ascorbic acid, vitamin E
We have already touched upon iron sequestration as a device for inhibiting microbial growth in section 17.5. A second benefit of keeping levels of free iron low is the prevention of •OH radical formation by Haber-Weiss chemistry (see slide 18.2.5). The free concentration of copper ions, which can react similarly, is kept low as well.
While metal sequestration reduces formation of reactive species, antioxidant enzymes and metabolites are needed to scavenge those that will form nevertheless. Glutathione has been known as a key antioxidant for a long time, but the great significance of the peroxiredoxins has only more recently been understood. These are enzymes that scavenge H2O2 and other peroxides by allowing their thiol-groups to be oxidized to disulfides. With both peroxiredoxins and glutathione, the disulfide forms are then reduced again at the expense of NADPH.
| 18.7.1 |
Iron chelation by heme and by transferrin |
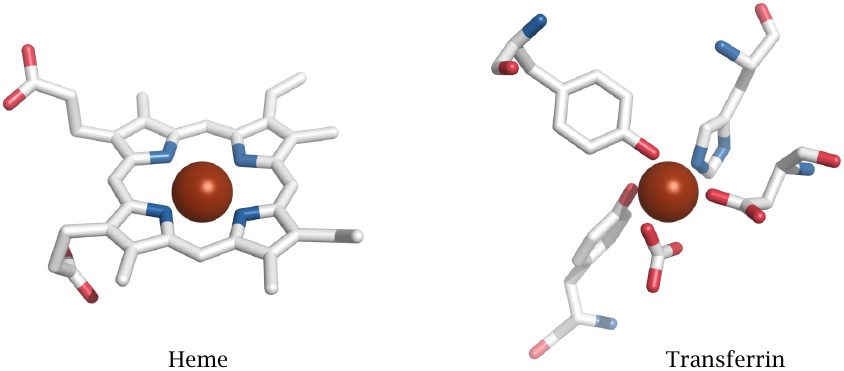
When it comes to preventing ROS formation, not all iron chelators are equally effective; depending on how many of the metal ion’s orbitals are engaged within the complex, its reactivity may be partially or completely suppressed. A case in point is heme, whose purpose is of course to maintain some degree of iron reactivity for oxygen binding, electron transport, or catalysis. The flip side is that heme can promote ROS formation, and as noted before, through association with lipid membranes or lipoproteins can facilitate lipid peroxidation. Therefore, accumulation of unbound heme must be prevented; and it is, through both binding proteins (hemopexin) and degradative enzymes (heme oxygenase).124
In contrast, transferrin, the major extracellular iron-binding protein, engages all outer orbitals of the iron ion and thereby renders it innocuous. Similarly, the iron-chelating drug desferrioxamine, which is used to treat hemochromatosis (see section 17.3.4) and other forms of iron overload, completely enwraps and neutralizes the cargo iron ion.125
| 18.7.2 |
Metallothioneins sequester copper and other heavy metals |
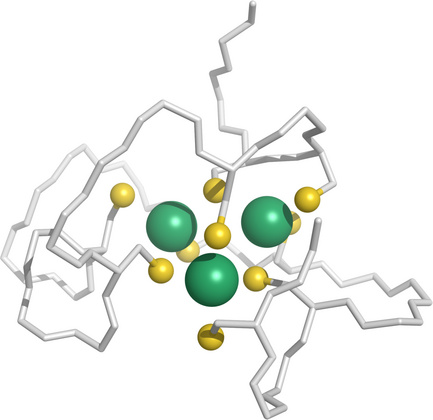
While the major extracellular transport protein for copper ions is albumin, intracellular copper is mostly bound to metallothioneins, which are small proteins that avidly bind copper, zinc, and various other metal ions. They provide a storage for copper and zinc while keeping their free concentrations low. There are at least four metallothionein (MT) isoforms that are expressed in humans, and which differ in metal affinity; for example, overexpression of MT-3, but not of MT-1, can starve cells of zinc to the point of killing them [186].
Metallothioneins also bind mercury and cadmium, which may mitigate the acute toxicity of these elements, although it would as such not promote their ultimate elimination.
This figure (rendered from 1j5l.pdb) shows three cadmium ions (green) bound to one of the two domains of a metallothionein from lobster, which enwraps the metal ions with a cluster of cysteine residues.
| 18.7.3 |
Superoxide dismutases contain transition metals |
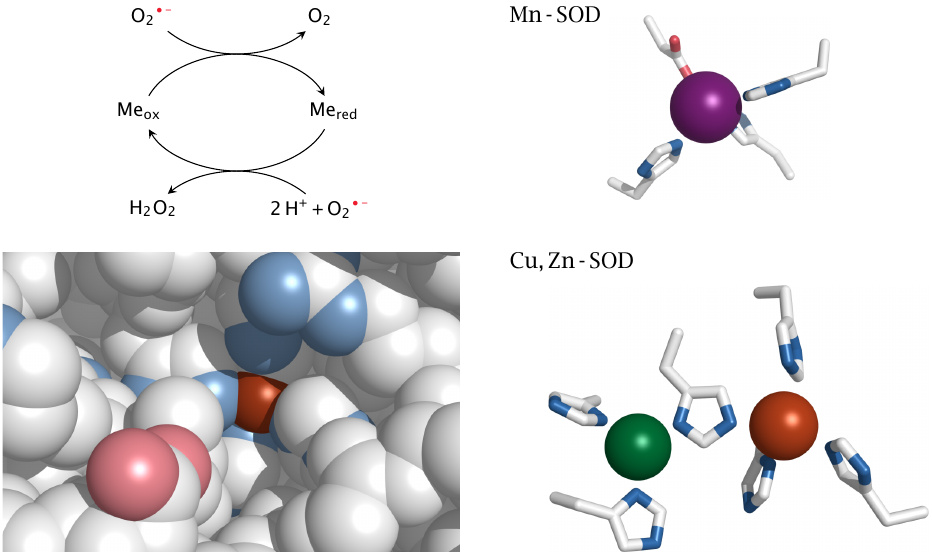
Superoxide dismutases react with O2•− radicals one at a time, which circumvents the problem of their mutual electrostatic repulsion (see footnote to slide 18.3.2). In doing so, a SOD molecule retains an electron from the first O2•− and donates it to the second. This electron is stored transiently on a transition metal ion; the chemical element employed for this purpose varies between different types of SOD. In the mitochondria, we find manganese-SOD, while in the cytosol and extracellularly we find copper, zinc-SOD; iron-containing SOD variants are common in bacteria.
For the two human enzymes, this slide shows how the metal ions are bound within their active sites by clusters of histidine residues (as well as one aspartate with Mn-SOD). In the case of Cu, Zn-SOD, the redox-active ion is copper (shown in brown), and only it, but not the zinc ion, is accessible to the substrate (see bottom left panel). One histidine residue sits between the two metal ions and presumably relays a modulating influence of Zn upon Cu.
Slide 18.2.5 showed that the first steps of both the SOD reaction and of the Haber-Weiss reaction consist in the reduction of a transition metal by O2•−. This raises the question how SOD avoids completing the second step of the Haber-Weiss reaction, namely the reduction of H2O2 to OH− and •OH. An interesting study on Cu, Zn-SOD [187] provides some insight into this. It turns out that the enzyme may indeed decompose H2O2 and thereby form a strong oxidant, presumably •OH; this oxidant, however, is not released but is instead retained on the catalytic copper ion. From there, it may be scavenged by various reducing agents, including uric acid, which will reactivate the enzyme; if this does not occur, it will destroy both itself and the enzyme by reacting with the His residue that is wedged between the Cu and Zn ions. Either way, the enzyme will avoid releasing the pernicious •OH into solution.
Structures rendered from 1cb4.pdb (Cu, Zn-SOD) and 2adq.pdb (Mn-SOD).
| 18.7.4 |
Structure of mitochondrial peroxiredoxin 3 |
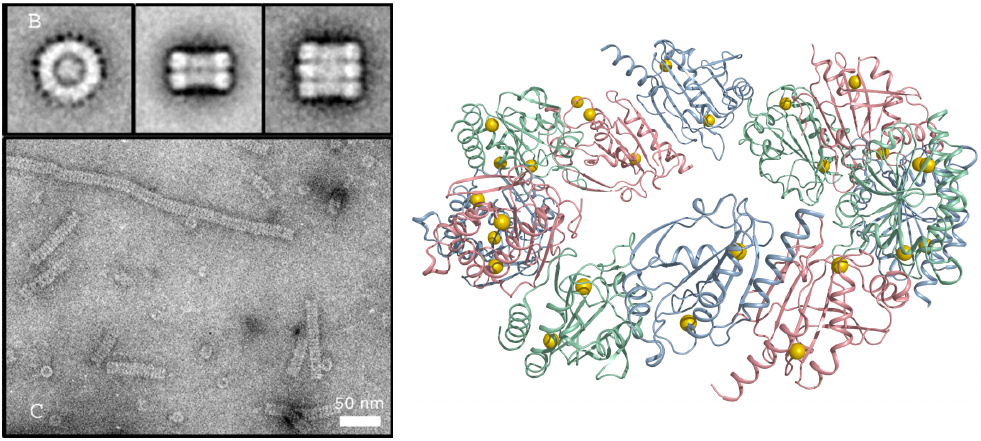
Like SODs, peroxiredoxins also occur in multiple variants that differ between cellular compartments. In keeping with the high rate of ROS production in the respiratory chain, they are particularly abundant in the mitochondrial matrix, where they account for up to 5% of total protein [188]; the contribution of peroxiredoxin to ROS detoxification in this compartment is discussed in slide 18.7.6.
A prominent mitochondrial variant is peroxiredoxin 3. It assembles into interesting oligomeric structures composed of stacked rings. Reaction with H2O2 induces disulfide formation between adjacent subunits. The molecular structure shown on the right represents the reduced state of one ring. Note that in this form the sulfhydryl groups (yellow balls) are not close to each other; therefore, a major conformational change must occur to permit their combination into disulfides. This conformational effect is thought to signal the change of redox state to other proteins.
Molecular structure rendered from 4mh2.pdb; electron microscopy taken from [189].
| 18.7.5 |
Other enzymes that carry out thiol/disulfide chemistry |
| Enzymes | Properties and functions |
| Glutathione peroxidases | contain selenocysteine in the active site; reduce organic peroxides |
| Thioredoxins | reduce protein disulfides, including peroxiredoxins |
| Thioredoxin reductase | reduces thioredoxin reductase using NADPH |
| Glutaredoxins | reduce protein/GSH mixed disulfides (P–SS–G) and dehydroascorbic acid |
| Thiol-disulfide isomerases | reside inside the ER; facilitate protein folding by resolving aberrant protein disulfides |
Most of the enzymes listed here contribute directly or indirectly to the scavenging of reactive oxygen species. Glutathione peroxidase is discussed in the next slide. Thioredoxins, glutaredoxins, and protein-disulfide isomerases all reduce protein disulfides, in different contexts as indicated. Glutaredoxins consume reduced glutathione; thioredoxins and protein thiol-disulfide isomerases initially form internal disulfides, which are then reduced by thioredoxin reductase using NADPH.
| 18.7.6 |
Detoxification of mitochondrial superoxide |
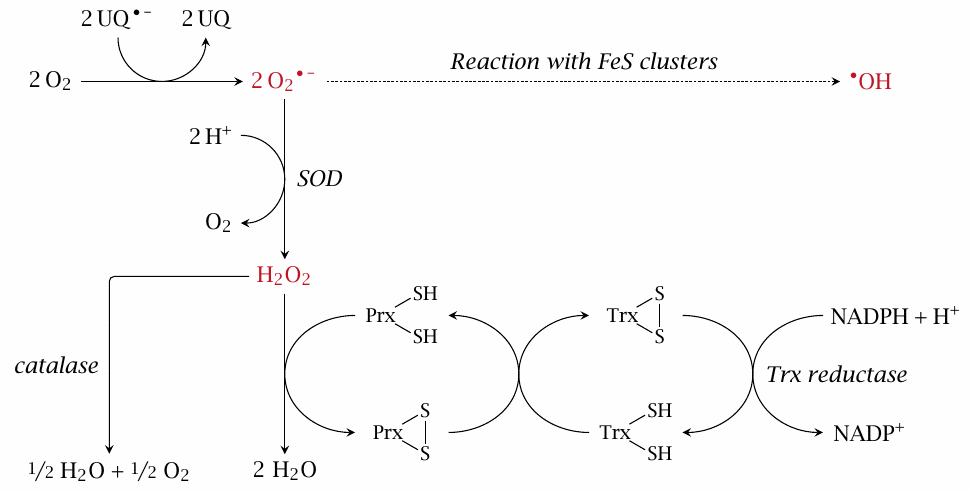
We had considered earlier how O2•− produced in the respiratory chain may yield •OH after reacting with iron-sulfur clusters (see slide 18.3.4). This slide illustrates how several antioxidant enzymes cooperate to prevent this. Most of the O2•− is scavenged by SOD, which disproportionates it to O2 and H2O2. The latter can be further disproportionated to water and O2 by catalase; however, this enzyme is not very abundant in mitochondria. Therefore, most H2O2 is instead reduced to water by peroxiredoxin (Prx), which is present at very high concentration. Reaction with H2O2 converts Prx to a disulfide, which is then reduced again by thioredoxin (Trx) and thioredoxin reductase.
| 18.7.7 |
Scavenging of organic peroxides by glutathione peroxidase |
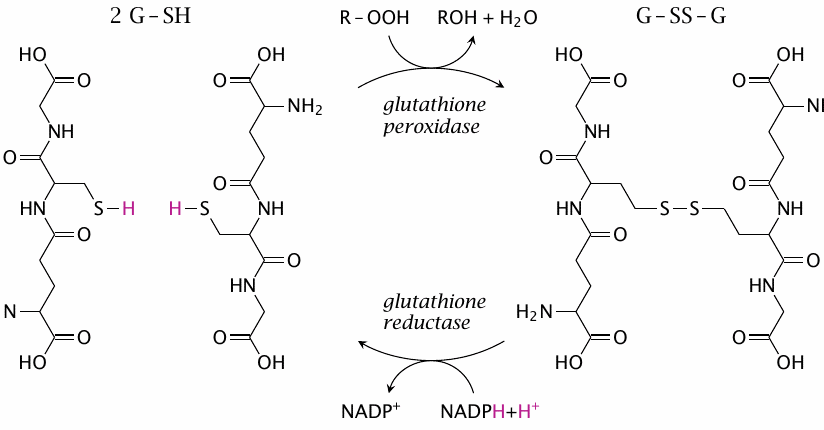
Glutathione peroxidases overlap in function with peroxiredoxins; they are, however, the main scavengers of organic peroxides (R–OOH), whereas H2O2 is scavenged mostly by peroxiredoxins. Interestingly, the active site of a glutathione peroxidase molecule contains a selenocysteine residue. The selenide group in its side chain (R–SeH), in its anionic form (R–Se−), reacts similarly to a cysteine thiolate (R–S−) but more rapidly. In the course of the reaction, the enzyme becomes transiently bound to glutathione (in the following form: R–Se–S–G). It is then reduced and released again by a second equivalent of glutathione; the reaction mechanism is analogous to that shown in slide 18.2.4.
As mentioned at the beginning of this chapter, glutathione is the most abundant antioxidant, and also a very versatile one; it can scavenge many radicals and channel their electrons to O2•− and SOD (see slide 18.2.3), and it contributes to the regeneration of other antioxidants. In particular, glutathione can reduce ascorbate, which in turn can regenerate urate and α-tocopherol. Through its reduction by the eponymous reductase and NADPH, glutathione taps into mainstream pathways of nutrient degradation and ensures a robust supply of reducing power for the scavenging of reactive species.
| 18.7.8 |
Ascorbic acid (vitamin C) is a major radical scavenger |
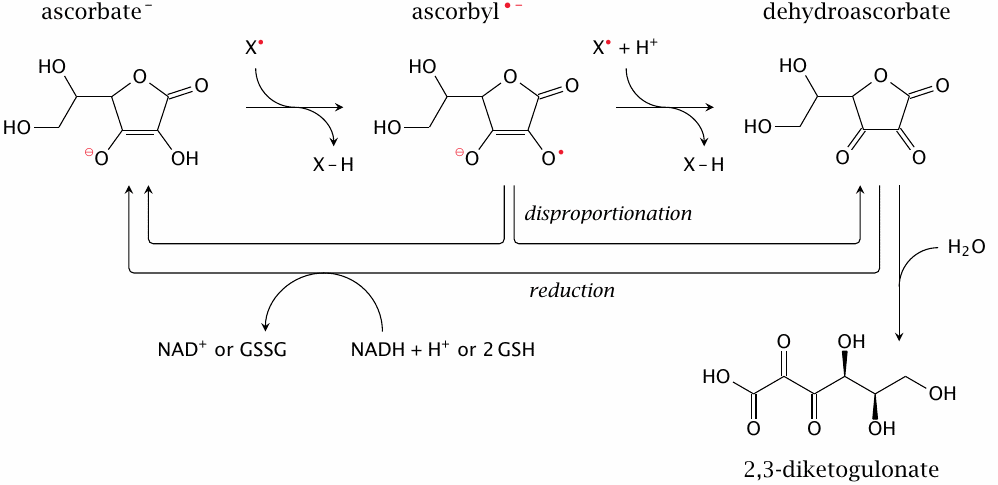
Ascorbate can scavenge radicals in two sequential reactions, although the second is less likely to occur than the first, since the free concentration of ascorbyl is kept low by disproportionation. It can be regenerated from dehydroascorbate using glutathione; this reaction is carried out by glutaredoxin. Since free glutathione and glutaredoxin levels are much higher inside the cell than outside, this pathway only occurs intracellularly.126 However, ascorbate scavenges radicals outside of cells, too. Extracellular dehydroascorbate can be reduced to ascorbate after uptake into the cell, which is facilitated by various glucose (GLUT) transporters [190,191]. An alternate pathway for reducing extracellular dehydroascorbate that does not require uptake into the cell is outlined in slide 18.7.14.
Dehydroascorbate that is not reduced may undergo non-enzymatic hydrolysis to 2,3-diketogulonate. In humans, ascorbate lost in this manner needs to be replaced from the diet. However, animals other than primates can produce ascorbate from glucose via d-glucuronate and l-gulonate and thus do not depend on the diet for ascorbate replenishment.
| 18.7.9 |
The energetics of ascorbyl disproportionation |
| Oxidised form | Reduced form | Δ E0′ (V) | |
| ascorbyl •− | −e− | dehydroascorbate | 0.174 |
| ascorbyl •−+H+ | +e− | ascorbate− | 0.282 |
| 2 ascorbyl•−+H+ | → | ascorbate− + dehydroascorbate | 0.454 |
The redox potentials for the reduction of dehydroascorbate to ascorbyl, and of the latter to ascorbate, were already given in slide 18.2.10. To determine the energy of ascorbyl disproportionation, we simply invert the first reaction and accordingly the sign of its potential. For the sum of both reactions, we obtain a strongly positive reaction potential, which equals a strongly negative free energy. Thus, even when reducing power for converting ascorbyl back to ascorbate is not immediately available, the equilibrium of disproportionation will keep the free concentration of ascorbyl very low.
If we also consider that ascorbyl is stabilized by resonance and therefore less reactive than most other radicals, it becomes clear that Nature made a very smart choice by picking ascorbic acid as its favorite radical scavenger.
| 18.7.10 |
Uric acid as a radical scavenger and antioxidant |
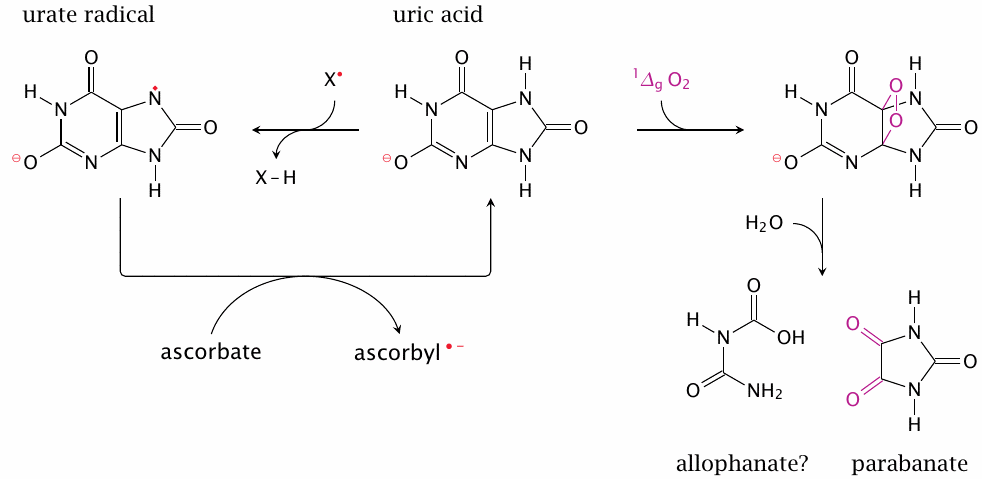
Uric acid is the final product of purine degradation in primates (see section 16.5), and it is subject to tubular reuptake in the kidneys, indicating that it constitutes a valuable commodity. It has indeed been shown that urate’s antioxidant effectiveness resembles that of ascorbate [163,192,193]. Since in primates the plasma concentration of urate exceeds that of ascorbate, it is fair to consider urate the major extracellular antioxidant.127
Uric acid may scavenge reactive species in two different ways. It may act as a radical scavenger (left); the urate radical formed in the process can then be reduced by ascorbic acid. Urate is also an effective scavenger of singlet oxygen; this reaction consumes urate and produces parabanate [195]. Allophanate as the other fragment seems plausible, but it may undergo further hydrolysis (as may parabanate). Urate can also react irreversibly with peroxynitrite and other reactive species [160].
| 18.7.11 |
α-Tocopherol intercepts lipid peroxidation |
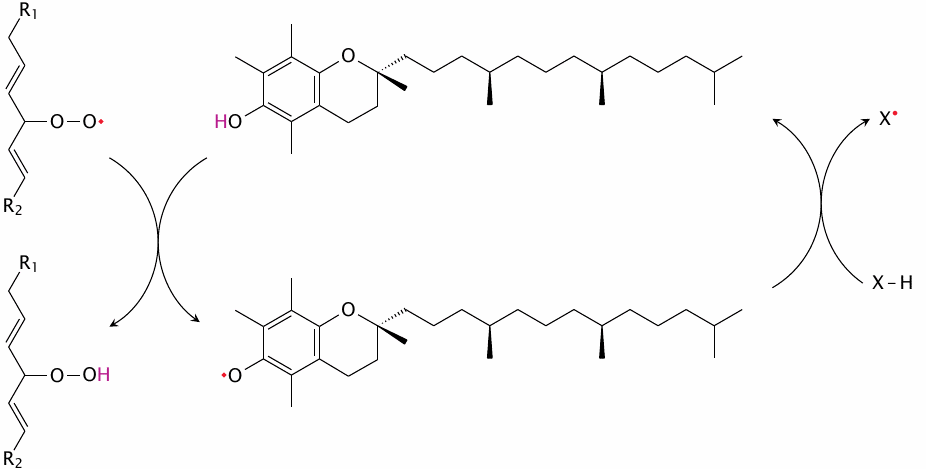
It was noted above that α-tocopherol, which is the most biologically effective constituent of vitamin E, plays a key role in keeping lipid peroxidation in check (see section 18.5).128 As is the case with other radical scavengers, the radical form of α-tocopherol is stabilized by resonance and therefore relatively benign. While it may react covalently with a second fatty acyl radical, more commonly it is regenerated at the expense of a secondary antioxidant (shown here simply as X–H). The most important secondary antioxidant is ubiquinol (see below).
| 18.7.12 |
Extracellular antioxidants |
- Small molecules: ascorbate, urate, glutathione
- Albumin
- Peroxiredoxin 4
- Selenoprotein P
The extracellular space contains much less antioxidant activity than is available inside cells. It also needs less; the respiratory chain and most other pathways that produce ROS occur only within cells. However, an important exception would be inflammation—as discussed above, phagocytes will release a brew of reactive species in order to kill extracellular pathogens, and in the process inflict damage on innocent bystander cells and molecules also.
Extracellular concentrations of ascorbate, urate, and glutathione are in the low hundreds of micromoles per liter. The regeneration of reduced ascorbate and urate is discussed in the next slide; oxidized glutathione cannot be regenerated outside the cell but is instead broken down into its constituent amino acids before cellular reuptake.
Albumin, the most abundant protein in the blood plasma, contains a free cysteine thiol that may react with radicals. The protein also binds copper and transports copper in the bloodstream. While albumin does not completely prevent the bound copper from engaging in Fenton chemistry, it reacts with and thereby scavenges the •OH thus produced, which prevents damage to other, less abundant and expendable target molecules.
While most peroxiredoxins are found intracellularly, peroxiredoxin 4 is secreted and may serve as an extracellular antioxidant. To do so effectively, there should be a pathway for its regeneration, for example by extracellular glutathione, but I have not been able to ascertain whether such a mechanism indeed exists.
Selenoprotein P is a unique protein that is rich in selenocysteine residues, and which serves both as an extracellular antioxidant and as a transport form of selenium [196].129
| 18.7.13 |
Regeneration of α-tocopherol by ubiquinol |
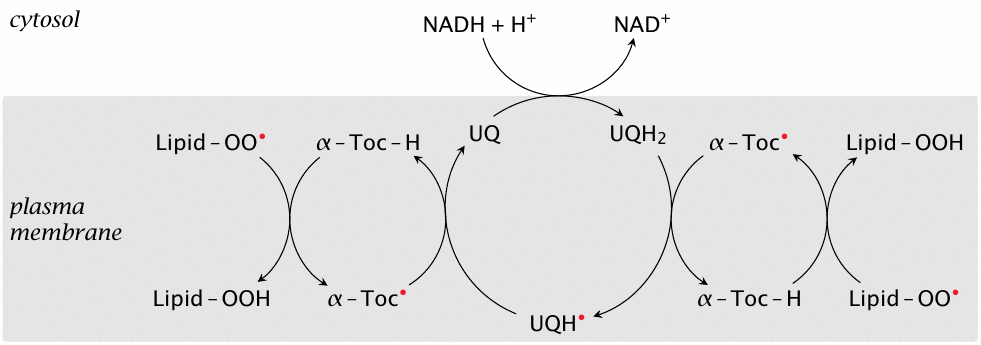
While ubiquinone (coenzyme Q) is well known for its role in the respiratory chain (see section 6.6.3), it is found not only in the inner mitochondrial membrane but also in all other cell membranes. Its main purpose there is to maintain α-tocopherol in a reduced state for continued control of lipid peroxidation, according to the pathway depicted in this slide.130
Ubiquinone itself is reduced in turn by NADH at the membrane interface. Since NADH is continually produced by glycolysis and other degradative pathways, this reaction provides the membrane with a robust supply of radical scavenging capacity. As the scheme shows, ubiquinone can accept electrons two at a time from NADH and then donate them singly to α-tocopherol. Single and pairwise transfer of electrons by ubiquinone is also observed in the respiratory chain (see section 6.6.3).
The reduction of ubiquinone by NADH is mediated by an enzyme known variously as diaphorase, methemoglobin reductase, and NADH-cytochrome b5 reductase [198]. We again note a low degree of substrate specificity in a reductive enzyme; this particular one reduces not only the substrates indicated in these different names, but also various drugs (see for example slide 19.5.3).
| 18.7.14 |
Regeneration of extracellular ascorbate and urate |
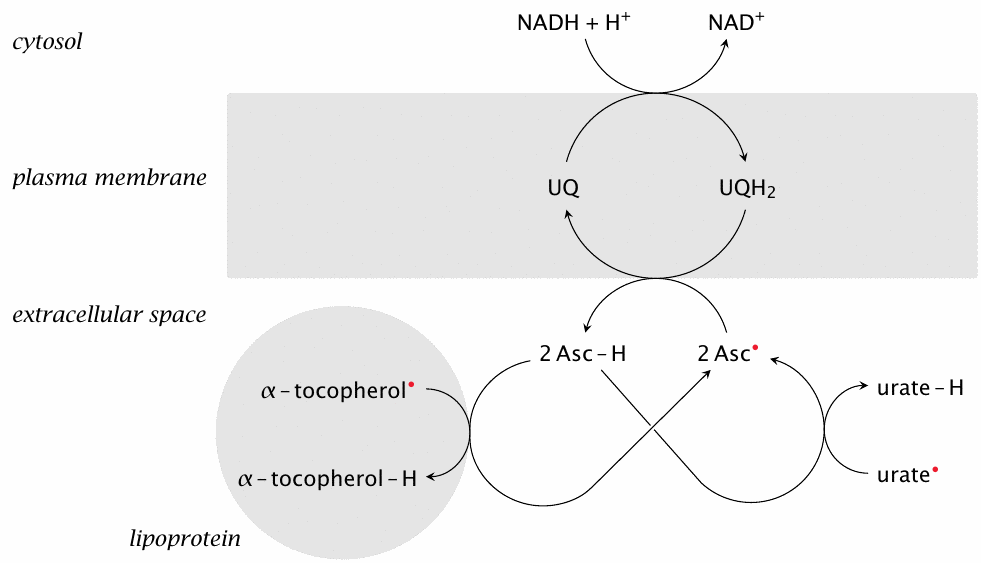
The NADH/ubiquinone pathway that sustains reduced α-tocopherol within cell membranes can also export reducing equivalents across them. Reduced ubiquinone (or ubiquinol) can reduce extracellular ascorbyl to ascorbate, which in turn reduces other extracellular radical scavengers, both hydrophilic (urate) and hydrophobic ones (α-tocopherol).
As noted in section 18.7.8, the cellular uptake of dehydroascorbate provides an alternate means for regenerating extracellular reducing power; in the bloodstream, this involves mostly the red blood cells. Uptake of dehydroascorbate via GLUT1 transporters into these cells is fast [190], but the release of reduced ascorbic acid seems to be slow; therefore, the ubiquinone-mediated regeneration of dehydroascorbate depicted here may be the predominant mechanism for the cellular export of reducing power.