| 4 |
Catabolism of sugars other than glucose |
| 4.1 |
Dietary sugars other than glucose |
| Trivial name | Composition | Source |
| lactose (milk sugar) | disaccharide of glucose and galactose | milk |
| sucrose | disaccharide of glucose and fructose | sugar cane, sugar beet, other fruits |
| fructose | monosaccharide | various fruits |
| sorbitol | sugar alcohol | fruits; semisynthetic |
| ribose, deoxyribose | monosaccharides | nucleic acids |
Starch is the most abundant carbohydrate in our diet, which makes glucose the most important dietary monosaccharide. However, our diet contains several other sugars in significant amounts. The guiding motif in the metabolism of these sugars is economy: instead of completely separate degradative pathways, there are short adapter pathways which merge into the main pathway of carbohydrate degradation, that is, glycolysis.
Lactose and sucrose are disaccharides. Degradation of both sugars begins with hydrolytic cleavage, which releases glucose and galactose or glucose and fructose, respectively. Fructose is also found in the diet as a monosaccharide. We already know how glucose is degraded, so we here only need to concern ourselves with the remaining monosaccharides. The degradation of sorbitol will be discussed as well, whereas ribose and deoxyribose will be covered in later chapters.
| 4.2 |
Degradation of fructose and sucrose |
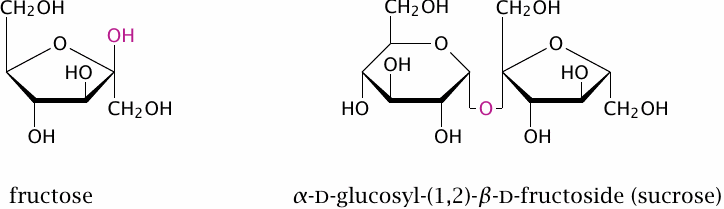
Sucrose is produced from sugar cane and sugar beet, which contain it in high concentrations (15–20%). In a typical Western diet, it may amount to as much as 20% of the total carbohydrate intake. Sucrose consists of glucose and fructose joined by a β-glycosidic bond between the carbon 1 of glucose and carbon 2 of fructose.
The hydrolytic cleavage of sucrose, like that of of maltose, occurs at the surface of the intestinal epithelial cells. The enzyme responsible is β-fructosidase, also named sucrase. Both sugars are then taken up by specific transport: Glucose by the SGLT1 transporter, and fructose by the GLUT5 transporter, which is named after glucose but actually transports fructose more effectively than glucose.
| 4.2.1 |
The fructolysis pathway |
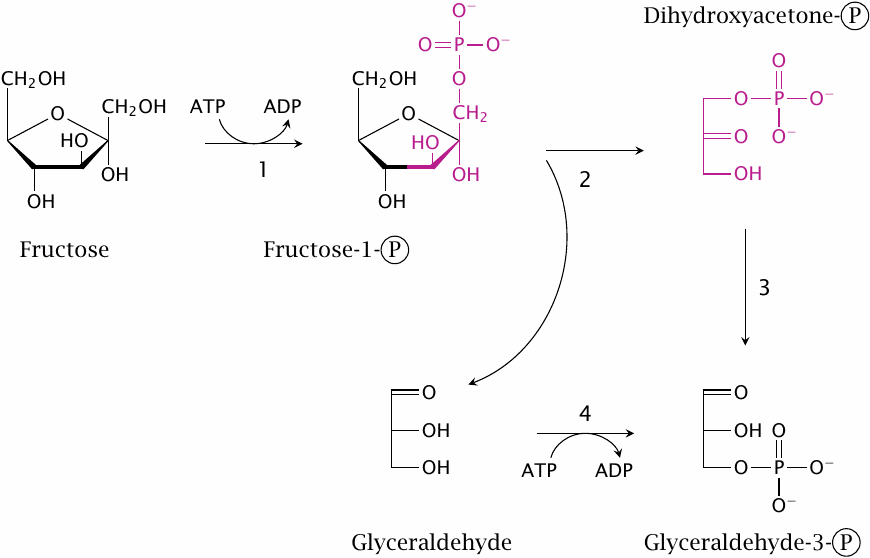
Fructose degradation, also called fructolysis, runs mostly in the liver. In the first step, fructose is phosphorylated by fructokinase (1), which uses ATP as a cosubstrate. This yields fructose-1-phosphate. The latter is then cleaved by aldolase B (2). The products of this reaction are dihydroxyacetone phosphate, which is already a metabolite in glycolysis, and glyceraldehyde, which can enter glycolysis after phosphorylation by glyceraldehyde kinase (4).
Glyceraldehyde can alternately be utilized by conversion to glycerol and then to glycerol-1-phosphate. The latter is a substrate in the synthesis of triacylglycerol, that is, fat. Fructose and sucrose appear to promote obesity more strongly than equivalent amounts of starch or glucose, and it has been suggested that its utilization via glycerol-1-phosphate, with subsequent triacylglycerol synthesis, may be among the reasons.
| 4.2.2 |
Fructose intolerance |
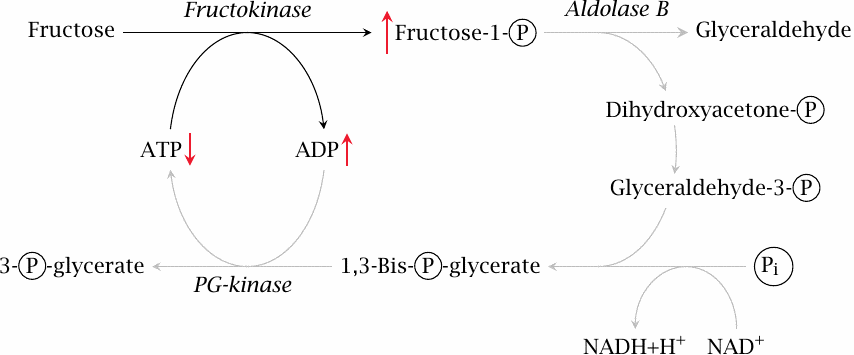
Fructose intolerance is a hereditary disease caused by a homozygous defect in the aldolase B gene. In this condition, fructose is still phosphorylated by fructokinase. The resulting fructose-1-phosphate, however, cannot be processed further, and therefore the phosphate tied up in it cannot be reclaimed. Since phosphate is required for the regeneration of ATP from ADP, this means that ATP will be lacking, too, which will sooner or later damage or even destroy the cell. Accordingly, the disease is characterized by potentially severe liver failure.
Fructose, alone or in combination with glucose, has been used in the past in the intravenous nutrition of intensive care patients; the perceived advantage of this treatment was the insulin-independent utilization of fructose. However, large intravenous dosages of fructose can significantly deplete liver ATP [8]; it appears that, under heavy load, aldolase B is unable to keep up with fructose kinase. Fructose is no longer a major component of intravenous nutrition schemes.
A defect in the gene encoding fructokinase leads to a condition named fructosemia or fructosuria. As these names suggest, fructose levels are increased both in the blood16 and the urine. Since fructose is not phosphorylated, no phosphate depletion occurs, and the liver cells do not incur any damage. The disease is therefore quite benign.
| 4.3 |
Lactose and galactose |
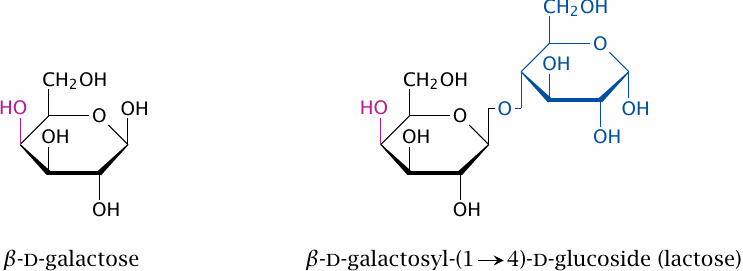
Lactose, a disaccharide of glucose and galactose, is the major carbohydrate contained in milk. Like maltose and sucrose, it is cleaved at the brush border of the small intestine, and the monosaccharide fragments are absorbed and passed along to the liver. The enzyme that accomplishes the cleavage is lactase or, more precisely, β-galactosidase.
| 4.3.1 |
The Leloir pathway for galactose utilization |
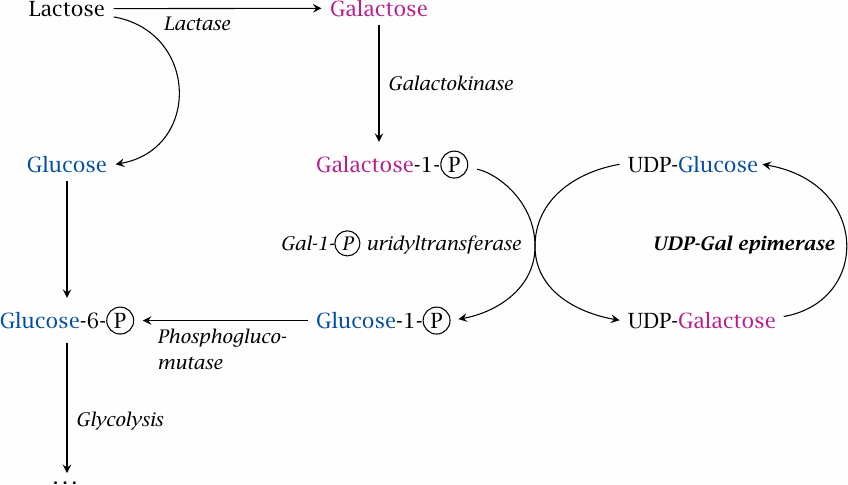
Galactose is utilized by conversion to glucose; this happens to a large extent in the liver, but the pathway is active in other tissues as well. The sugar is first phosphorylated by galactokinase. The resulting galactose-1-phosphate undergoes an exchange reaction with UDP-glucose, which is catalyzed by galactose-1-phosphate uridyltransferase and releases glucose-1-phosphate and UDP-galactose. Glucose-1-phosphate can be converted by phosphoglucomutase to glucose-6-phosphate, which is the first intermediate in glycolysis. UDP-galactose is converted to UDP-glucose by UDP-galactose epimerase.
In this pathway, UDP-glucose and UDP-galactose fulfill catalytic roles but are not subject to any net turnover, much like the intermediates in the citric acid cycle. It might therefore be said that they form a tiny metabolic cycle between the two of them. Also note that, save for the final epimerase reaction, the pathway is really just smoke and mirrors—performing the epimerization on galactose directly would accomplish the same net effect, without being chemically more difficult in any way.17
| 4.3.2 |
Mechanism of UDP-galactose epimerase |
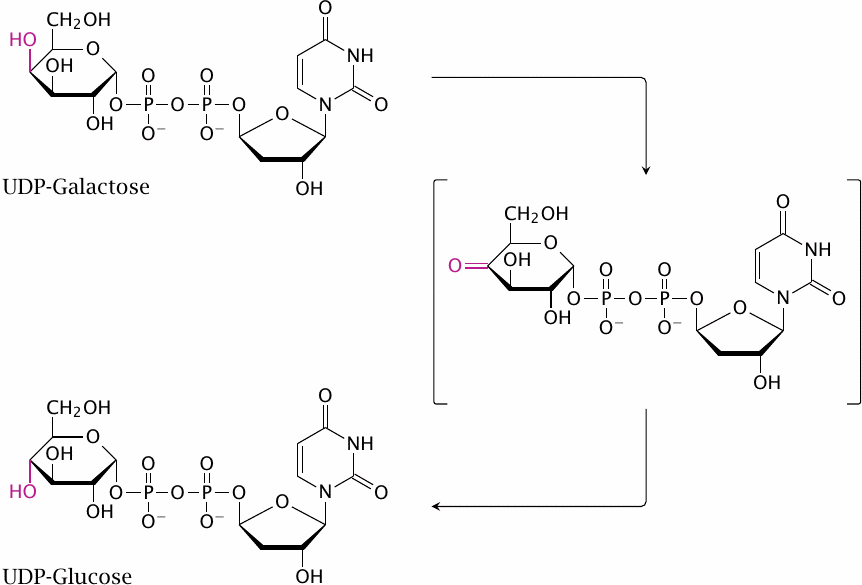
One unusual feature of UDP-galactose epimerase is its use of NAD+ as a coenzyme, rather than as a cosubstrate—that is, NAD+ undergoes no net reduction or oxidation in this case. Nevertheless, it functions here much in the same way as it does in other enzyme reactions. The 4′-hydroxyl group of the sugar is transiently dehydrogenated to a keto group, and in this step NAD+ accepts the abstracted hydrogen. The substrate is then rotated within the active site before the H2 is transferred back to it, which causes the 4′-OH group to now point the other way.
| 4.3.3 |
Lactose intolerance |
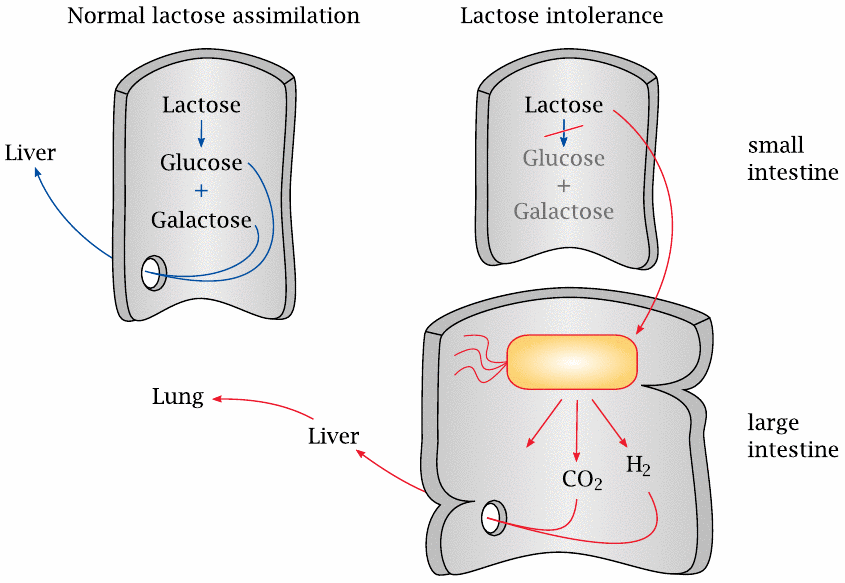
A deficiency of the lactase enzyme in the small intestine gives rise to lactose intolerance, which is found frequently in people of East Asian descent who are past their infant age.
If lactose is not cleaved, it cannot be absorbed, so it travels down the drain from the small to the large intestine. Many of the bacteria found there have the capacity to metabolize lactose, which they will happily convert to acids and gas. For example, Escherichia coli has a pathway called mixed acid fermentation. One of the products of this fermentation is formic acid (HCOOH), which is then cleaved by formic acid lyase to H2 and CO2. The decarboxylation of formic acid serves the same purpose as that of pyruvate in ethanolic fermentation, namely, the removal of excess acidity resulting from the fermentation (see slide 3.4.3).
The aberrant fermentation and gas formation leads to abdominal discomfort and diarrhea. Since the environment in the large intestine lacks oxygen, H2 generated in the bacterial fermentation is not oxidized but instead enters the system as such and is mostly exhaled. An increase in exhaled hydrogen gas provoked by ingesting a test dose of lactose can be used to diagnose the condition.
Treatment consists in omission of lactose in the diet. Milk can be pre-treated with purified bacterial β-galactosidase, rendering it suitable for consumption by lactose-intolerant individuals. Fermented milk products such as yogurt and cheese are depleted of lactose by microbial fermentation and therefore do not pose a problem for lactose-intolerant individuals.
| 4.3.4 |
Galactosemia |
| Type | Enzyme deficiency | Accumulating metabolites |
| I | galactose-1-phosphate-uridyltransferase | galactose, galactose-1-phosphate, galactitol, galactonate |
| II | galactokinase | galactose, galactitol |
| III | UDP-galactose epimerase | galactose-1 phosphate, UDP-galactose |
Three different enzyme deficiencies in the pathway are subsumed under the name galactosemia, which means “galactose in the blood.” All of these are rare; type I is the most common and most severe form. Here, the deficient enzyme is galactose-1-phosphate uridyltransferase. This leads to a buildup of galactose-phosphate, but also of several other metabolites. The disease becomes manifest in newborns with acute liver failure and is deadly if not promptly diagnosed and treated. In many countries, this enzyme defect is part of neonatal screening programs.
Therapy consists in the removal of galactose from the diet, but even so organ damage develops, most commonly affecting the CNS and, in girls, the ovaries. The residual pathology that develops in spite of the diet is ascribed to the endogenous synthesis of galactose, which proceeds via UDP-glucose and UDP-galactose; the UDP-galactose epimerase reaction is reversible.
For a long time, it was assumed that accumulation of galactose-1-phosphate and phosphate depletion are responsible for cell and organ damage, which is analogous to the pathogenic mechanism in fructose intolerance (see slide 4.2.2). However, this assumption has been called into question by the results of animal experiments. When galactose-1-uridyltransferase is genetically knocked out in mice, these develop a profile of metabolite accumulation that closely resembles human patients, but they do not display any of the pathology typically observed in humans [9]. What is more, some rare human cases have been reported that show the usual biochemical manifestations, but no clinical signs [10]. The quest for the true cause of the pathology affecting most human patients continues [11,12].
In the order of the pathway, type II galactosemia comes first, as it involves a defect of galactokinase. In this case, galactose simply does not enter the Leloir pathway at all; it builds up in the blood and is mostly eliminated in the urine. The liver will not be adversely affected. However, there is a common complication elsewhere: the eyes will develop cataract, that is, obfuscation of the lenses. This is due to the reduction of galactose to galactitol in the cells of these organs by aldose reductase (see slide 4.4).
The rarest form of galactosemia is due to the defect of UDP-galactose epimerase. The biochemical pattern is similar to type I, except that UDP-galactose also accumulates, and as in type I, developmental delay seems to occur [13]. In this condition, both the utilization and the synthesis of galactose are inhibited, and it appears necessary to maintain a low level of dietary galactose to supply the synthesis of galactose-containing glycolipids and glycoproteins.
| 4.4 |
Sorbitol is an intermediate of the polyol pathway |
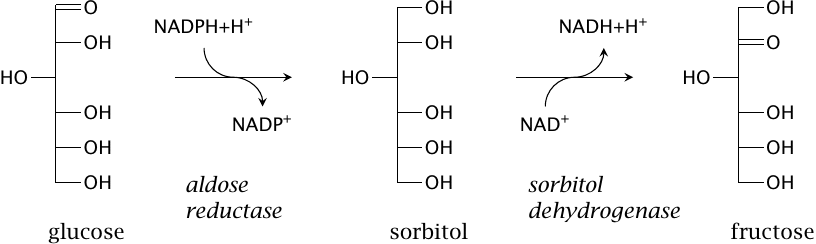
Sorbitol is not strictly a sugar but rather a sugar alcohol, since it has no keto or aldehyde group. It is normally a minor component of dietary carbohydrates, but it is also prepared semisynthetically and used as a sweetener. In addition, it is formed in our own metabolism from glucose in the polyol pathway, which then converts it further to fructose. Note the use of NADPH as a cosubstrate in the first step, and of NAD+ in the second. NADP exists mostly in the reduced state, while NAD is mostly oxidized (see slide 9.3.1), which means that both drive the pathway in the indicated direction.
The first enzyme, aldose reductase, is not specific for glucose but can also reduce galactose, which gives rise to galactitol. As stated above, the elevated blood level of galactose that occurs in galactosemia type II causes galactitol to accumulate in the lenses; the same occurs with glucose and sorbitol in insufficiently treated diabetes mellitus (see slide 14.5.7). Accumulation of either sorbitol or galactitol causes cataract; this is ascribed to their osmotic activity, which causes cell damage through swelling.
Like the cells in the lens, nerve cells are able to take up glucose in an insulin-independent fashion, and like cataract, nerve cell damage (diabetic polyneuropathy) is a common long-term complication in diabetes. It appears plausible that sorbitol accumulation might also be responsible for this nerve cell damage. Inhibitors of aldose reductase have been developed and have shown promise in animal models of both diabetes and galactosemia, but evidence of clinical effectiveness in humans is scarce.
Conversion of glucose to fructose via the polyol pathway occurs in the seminal vesicles, which are part of the male sexual organs, and fructose is found in the sperm fluid. It supplies the sperm cells with fuel in their frantic quest for an oocyte; the advantage of this somewhat unusual source of energy may be that fructose will not be pilfered by the other tissues the sperm fluid will get into contact with.
Now, if sperm cells require fructose to sustain their motility, one might expect that prevention of fructose synthesis with aldose reductase inhibitors would disrupt male fertility and thus might provide the long-sought pill for males. However, I have not seen any experimental studies on this subject.