| 15 |
Biosynthetic pathways using tetrahydrofolate and vitamin B12 |
| 15.1 |
Overview |
The transfer of single carbon units, in various redox states, is important in several biosynthetic pathways. The two coenzymes tetrahydrofolate and methylcobalamin play central roles in these reactions. Both coenzymes are derived from vitamins, and deficiencies of these vitamins are not uncommonly encountered in clinical medicine, usually as a consequence of another underlying medical condition.
We will first look at the biochemical reaction cycles and then consider the pathological consequences that result from their disruption through vitamin deficiencies.
| 15.2 |
Tetrahydrofolate-mediated carbon transfer reactions |
| 15.2.1 |
The role of tetrahydrofolate in biosynthetic reactions |
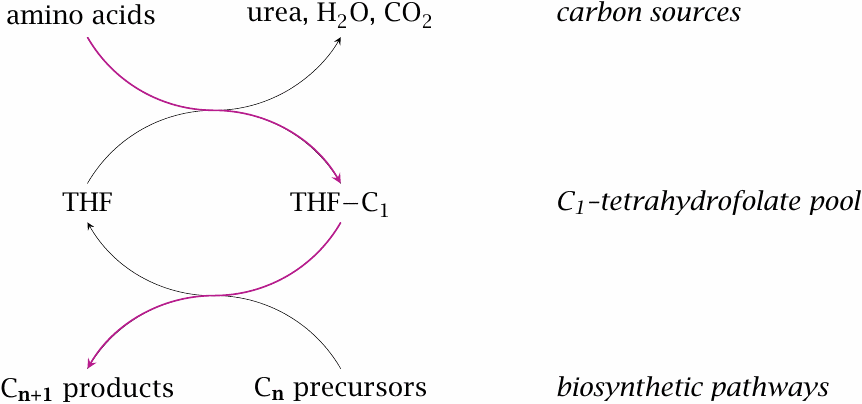
The degradation pathways for several amino acids produce single carbon units that are transferred to tetrahydrofolate (THF) as an intermediate carrier. Tetrahydrofolate, in turn, donates the single carbons to biosynthetic intermediates in various pathways.
| 15.2.2 |
Folic acid is reduced by dihydrofolate reductase (DHFR) |
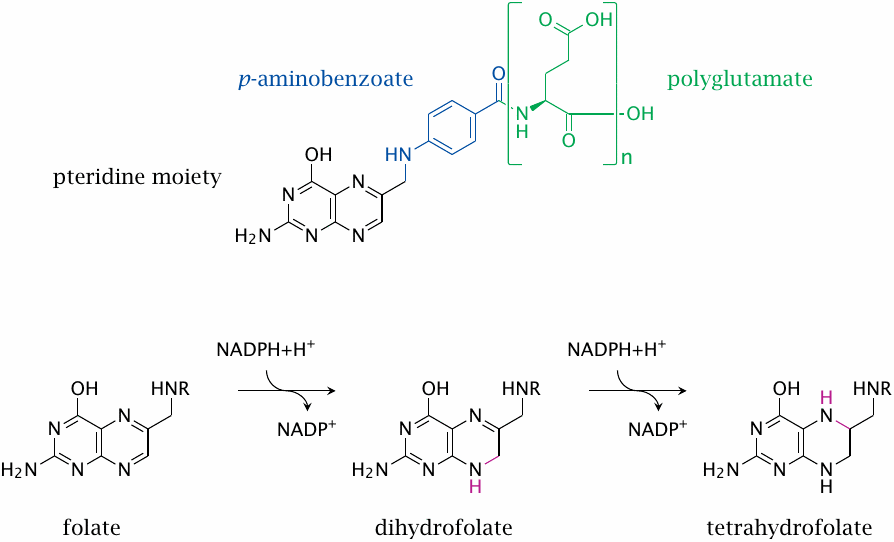
Folic acid is the vitamin as it is found in the diet. It consists of three distinct moieties: a pteridine moiety, a p-aminobenzoate moiety, and one or several glutamate residues.
The metabolically active form is tetrahydrofolate, which is formed from folate in two successive NADPH-dependent reductions, both catalyzed by the same enzyme, namely, dihydrofolate reductase.
The number of glutamate residues contained in folic acid or tetrahydrofolate (THF) changes at various stages of transport and utilization. Intestinal uptake and transport through the blood occur with only one glutamate attached. After uptake into liver cells, several more glutamate residues are added; and it is the polyglutamate form that functions as the actual cosubstrate. The exact number of glutamate residues present appears to be involved in the regulation of folate-dependent metabolism [100], but we are not going to consider this aspect in detail.
| 15.2.3 |
Sources and destinations of C1 units transferred by tetrahydrofolic acid |
Sources:
- serine, glycine
- histidine, tryptophan
Biosynthetic destinations:
- purine bases
- thymine
- S-adenosylmethionine → choline phospholipids, creatine, epinephrine, DNA methylation
Degradation of the amino acids glycine, serine, histidine and tryptophan provides C1 units to tetrahydrofolate. Serine and glycine are more abundant than the two others and therefore are the main sources.
| 15.2.4 |
The serine hydroxymethyltransferase reaction: release of CH2O |
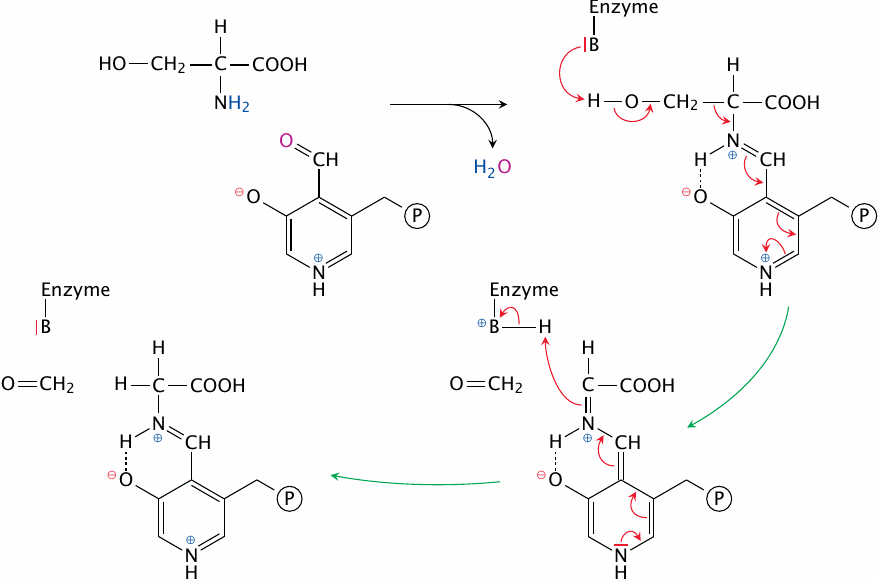
One of the major reactions that supply C1 units to THF is the cleavage of serine by serine hydroxymethyltransferase. The active site of this enzyme contains the coenzyme pyridoxal phosphate (PLP) and a catalytic base (here represented by B). As in amino acid transamination (section 12.2), the α-amino group of the substrate serine forms a Schiff base with PLP. Through the imine (C=N) bond, PLP can reversibly withdraw and donate electrons, which in cooperation with the catalytic base facilitates cleavage of the bond between the α- and the β-carbon. The β-carbon is released as formaldehyde, and the aldimine (Schiff base) is subsequently hydrolysed to release glycine (not shown).
Note that the whole reaction is reversible and therefore can bring about the synthesis of serine from glycine and N,N’-methylene--THF.
| 15.2.5 |
The serine hydroxymethyltransferase reaction (2): capture of formaldehyde by THF yields N,N′-methylene-THF |
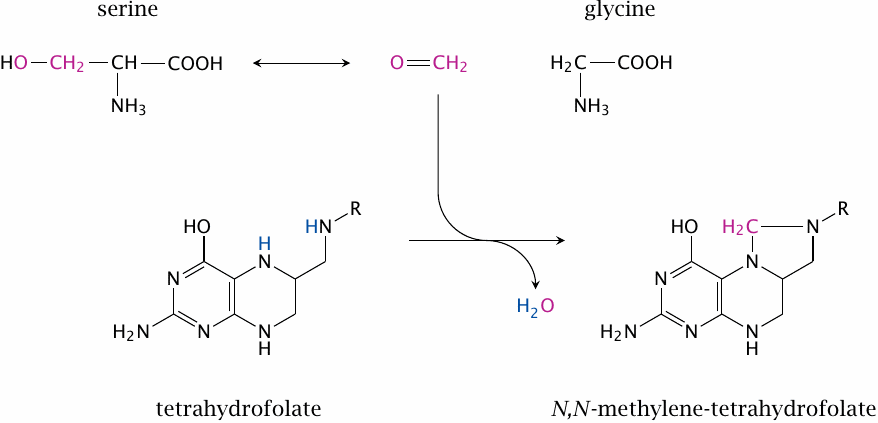
The formaldehyde released by serine cleavage is captured immediately by THF while still within the active site. This yields N,N’-methylene--THF.
It should be noted that, while the catalytic mechanism presented here is found in many textbooks, an alternate mechanism has been proposed in which THF participates more directly in the cleavage of serine, and free formaldehyde does not occur as an intermediate [101].
| 15.2.6 |
N,N′-methylene-THF production by the glycine cleavage system |
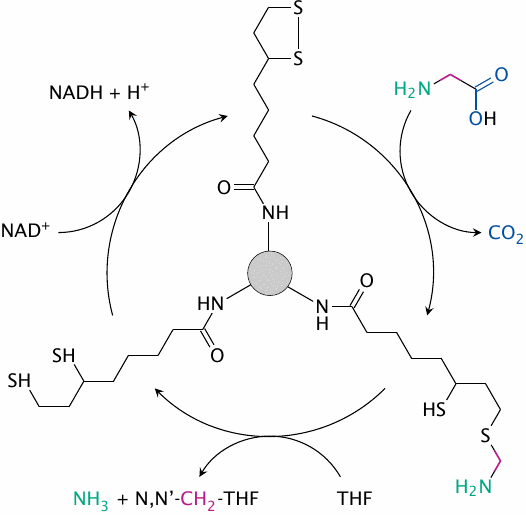
Another major reaction to supply C1 units to THF is the degradation of glycine by the glycine cleavage system. This is a multi-enzyme complex with three separate catalytic subunits and one substrate carrier protein. The latter possesses a lipoamide moiety to which the substrate becomes transiently attached, a feature that is also found in pyruvate dehydrogenase (see slide 5.2.7). The initial decarboxylation reaction uses pyridoxal phosphate as a coenzyme, and the second reaction again appears to involve the capture of a formaldehyde intermediate by THF.
In the third reaction, the dihydrolipoamide formed in the second step is dehydrogenated, which restores the initial disulfide form of lipoamide. This reaction is identical to the final steps in the pyruvate dehydrogenase, α-ketoglutarate dehydrogenase, and branched chain α-keto acid dehydrogenase reactions. Indeed, all four multienzyme complexes employ the same dihydrolipoate dehydrogenase subunit for this purpose (and moreover, they are all found in the mitochondria).
| 15.2.7 |
Histidine degradation produces N,N′-methenyl-THF |
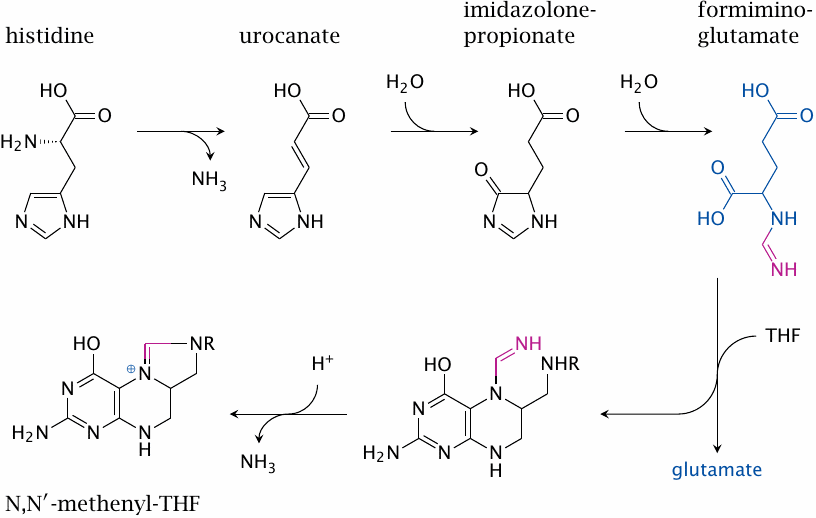
Degradation of histidine proceeds via the intermediates urocanate, imidazolone-propionate and formimino-glutamate. The formimine (or methenimine) group of the latter is transferred to tetrahydrofolate. Subsequent cleavage of ammonia yields N,N′-methenyl-THF, which can either be oxidized to formyl-THF or reduced to N,N’-methylene-tetrahydrofolate (see slide 15.2.8).
| 15.2.8 |
Redox transitions between various forms of C1-THF |
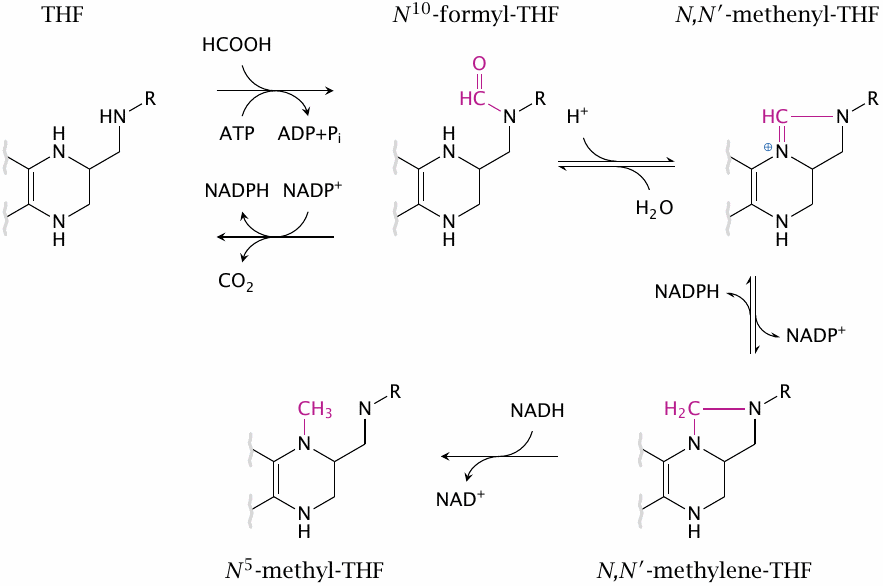
C1 derivatives of THF exist in different redox states that can be converted into one another. The final reduction, which leads from N,N’-methylene-tetrahydrofolate to N5-methyl-THF, is apparently irreversible; this is relevant for the methyl trap mechanism that accounts for the pathogenesis of anemia in vitamin B12 deficiency (see slide 15.5.6).
N10-Formyl-THF can be formed from THF through capture of formic acid, which in turn arises from tryptophan degradation. It can also be converted back to THF by NADP+-dependent dehydrogenation of the formyl group to CO2. The latter reaction is used to dispose of excess C1-intermediates when supply exceeds demand.
| 15.2.9 |
Overview of flux through the C1-THF pool |
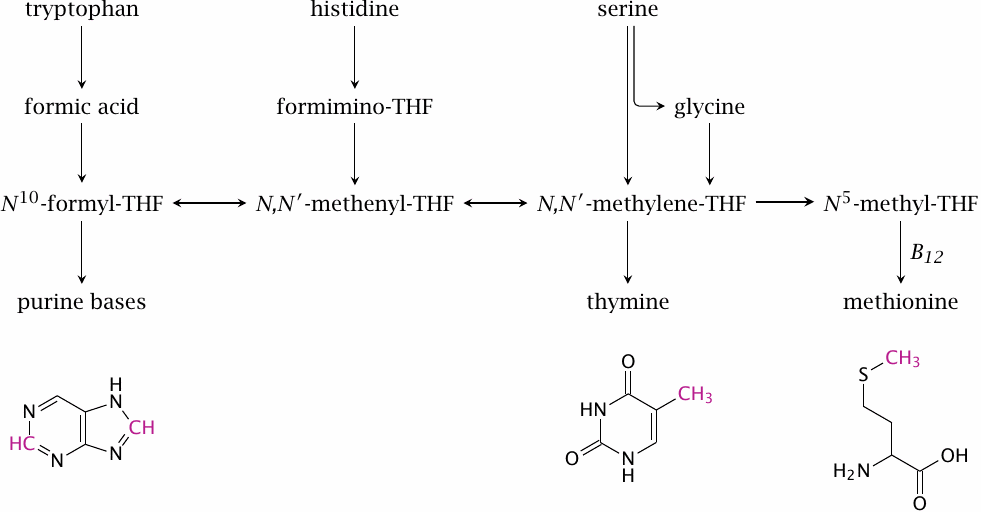
Formyl-THF and N,N’-methylene--THF donate single-carbon units in nucleotide synthesis. Methyl-THF donates methyl groups to the synthesis of methionine. Methionine synthesis constitutes the link between folate-dependent and vitamin B12-dependent C1 metabolism. An activated form of methionine functions as a methyl donor in several important biosynthetic pathways (see slide 15.4.2).
In the depicted structures of adenine, thymine and methionine, the carbons that are acquired via THF are highlighted.
| 15.3 |
Folate antimetabolites as antimicrobials |
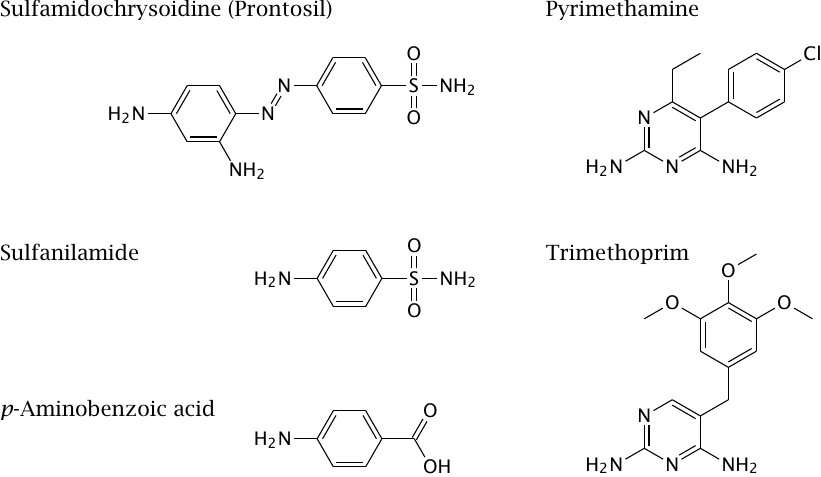
Folic acid metabolism in microbes contains drug targets for antimicrobial therapy. An important example are the sulfonamides, which became available in the 1930s and were the first broad-spectrum antibacterial drugs. The first such drug, sulfamidochrysoidine (trade name Prontosil), was developed as a red dyestuff. Its antibacterial activity was discovered in a test program that systematically evaluated the effects of all the company’s new compounds in animal experiments.
The compound as such, when tested in vitro, has no antibacterial activity. The active component is sulfanilamide, which is released in animals or humans by reductive metabolism. Its antibacterial action is due to its competition with p-aminobenzoic acid, which is a precursor that becomes incorporated into folic acid in its bacterial biosynthesis (see slide 15.2.2). Since humans do not synthesize folate, they are not affected by sulfanilamide.
The antibacterial effect of sulfonamides can be substantially increased by combination with inhibitors of dihydrofolate reductase such as trimethoprim. In this case, the selective toxicity of the inhibitor for bacteria is due not to the absence of the enzyme in humans—dihydrofolate reductase is needed in both bacterial and human metabolism—but instead to the molecular differences between the bacterial and the human enzyme.
Combinations of a sulfonamide and a dihydrofolate reductase inhibitor can also be used in the treatment of some protozoal infections. One such combination, sulfadoxine plus pyrimethamine, was widely used against malaria at one time, but the development of resistance among the malaria parasites has since made it obsolete in this application. It is still used against toxoplasmosis and Pneumocystis carinii infections.
Inhibitors of human dihydrofolate reductase, such as methotrexate, are used in the treatment of tumors and autoimmune diseases (see slide 16.9.5).
| 15.4 |
Cobalamin-dependent methylation reactions |
| 15.4.1 |
Structure of methylcobalamin |
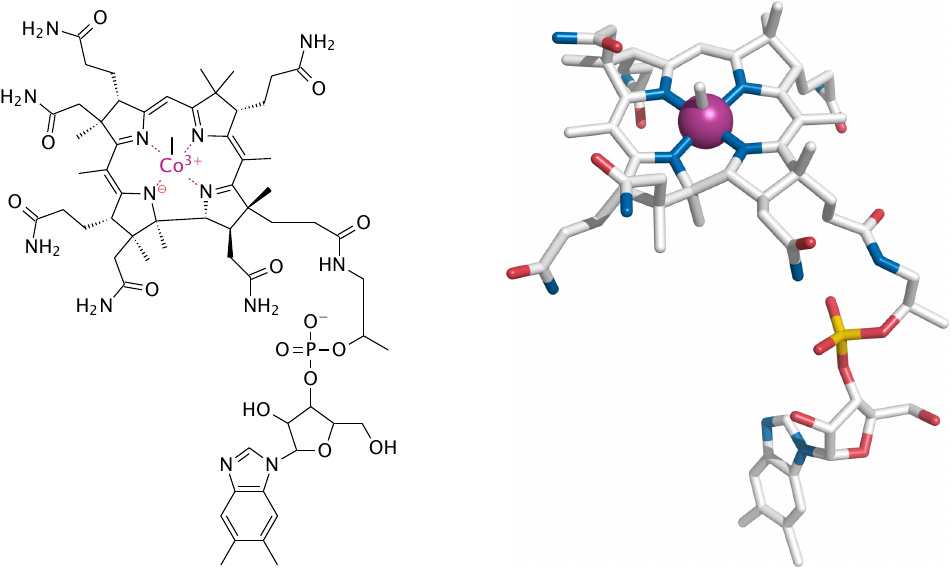
Cobalamin is a coenzyme with a complex structure. Its corrin ring coordinates a single cobalt ion, which is central to the ability of cobalamin to facilitate the cleavage and formation of carbon bonds by way of radical intermediates.
The two major forms of cobalamin are adenosylcobalamin and methylcobalamin. Shown here is methylcobalamin, which is formed as an intermediate in the synthesis of methionine from homocysteine (see below). The methyl group is obtained from N5-methyl-THF. In this chapter, we only deal with methylcobalamin-dependent reactions. Adenosylcobalamin is needed in the utilization of propionyl-CoA (see slide 10.3.6); it is not considered in detail in these notes.
| 15.4.2 |
The S-adenosylmethionine (SAM) cycle requires vitamin B12 |
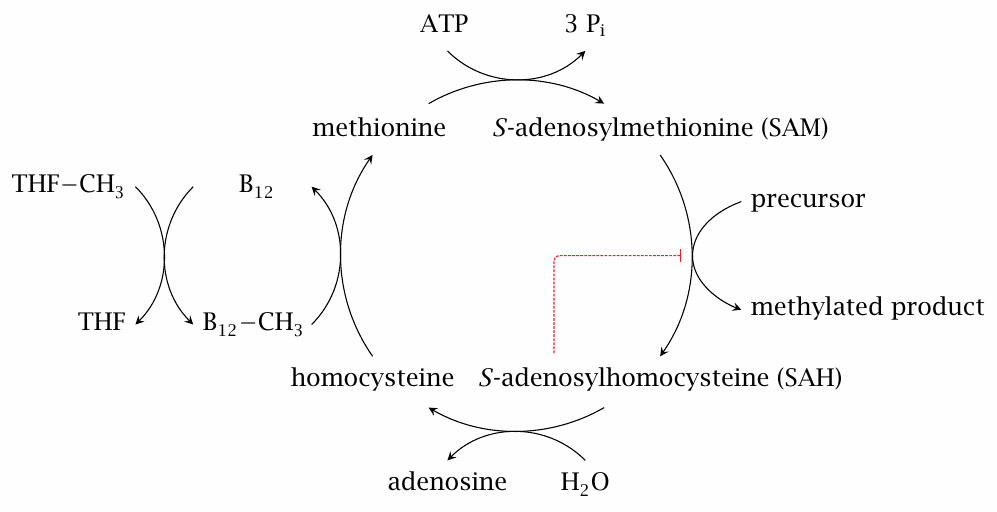
In the SAM cycle, methionine is first activated to S-adenosylmethionine (SAM), which then serves as a methyl group donor in various biosynthetic reactions. Methyl group donation leaves behind S-adenosylhomocysteine, which is then cleaved to adenosine and homocysteine. The regeneration of methionine from homocysteine requires vitamin B12 in the form of methylcobalamin.
S-adenosylhomocysteine (SAH) is not only the byproduct of SAM-dependent methylation reactions but also a competitive inhibitor. It must therefore be recycled promptly in order to avoid disruption of the SAM cycle. In vitamin B12 deficiency, the regeneration of SAM from SAH is impeded, and the accumulating SAH interferes with subsequent methylation reactions [102].
| 15.4.3 |
Structures of S-adenosylmethionine and S-adenosylhomocysteine |
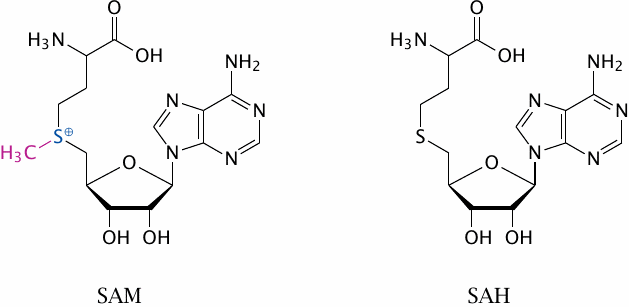
The key feature of SAM is the methyl group attached to a sulfonium ion (S+). The inherent instability of the sulfonium group makes this compound a good methyl group donor.
| 15.4.4 |
SAM-dependent methylation reactions |
- methylation of phosphatidylethanolamine (PE) to phosphatidylcholine (PC)
- guanidinoacetate → creatine
- norepinephrine → epinephrine
- acetylserotonin → melatonin
- DNA methylation
- methylation of drugs (e.g. mercaptopurine)
Among these pathways, the biosynthesis of PC and of creatine are quantitatively the most important ones.
| 15.4.5 |
Phosphatidylethanolamine methylation |
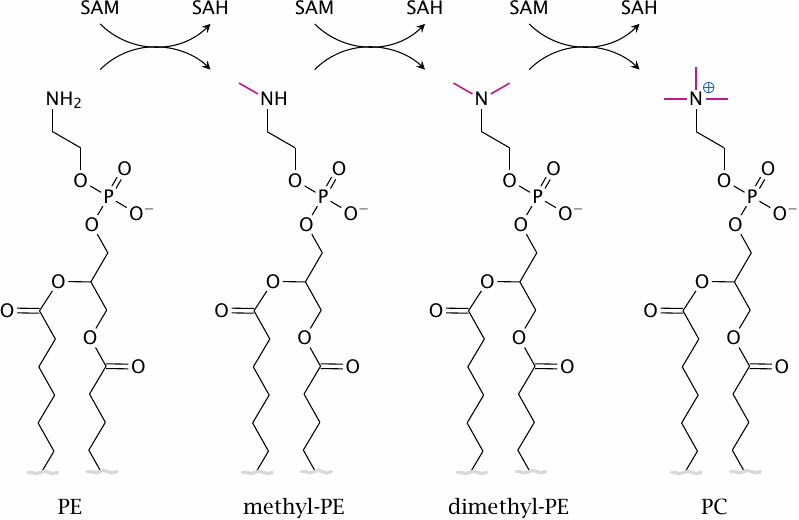
Phosphatidylcholine (PC) is a major constituent of mammalian cell membranes. It is formed in three successive SAM-dependent methylation steps from phosphatidylethanolamine (PE).
PC can also be synthesized using dietary choline, and conceivably this pathway might supply enough PC for myelin synthesis. However, in animal experiments, depletion of vitamin B12 has been shown to decrease PC and to increase PE in the brain [103], suggesting that PE methylation is important for sufficient PC synthesis at least in this organ.
| 15.4.6 |
Sphingomyelin acquires its phosphocholine headgroup from PC |
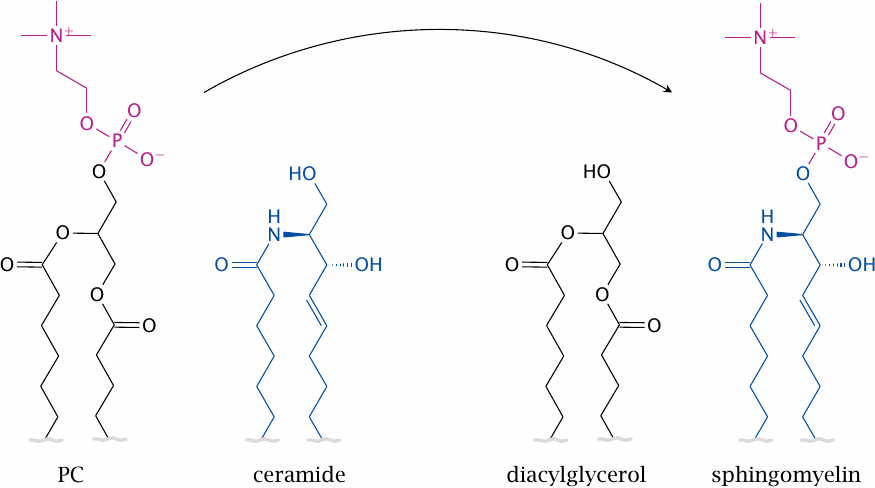
Sphingomyelin, another important membrane phospholipid, is synthesized from its precursor ceramide through phosphocholine transfer from PC; therefore, its synthesis indirectly also depends on the SAM cycle and vitamin B12.
| 15.4.7 |
Major nerve fibers are myelinated |
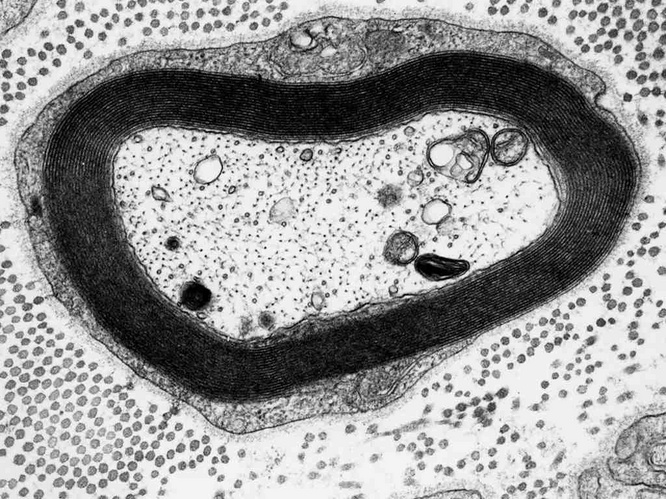
Myelin sheaths consist of multiple layers of cell membranes, which contain more phospholipids and less protein than most other cell membranes. These elaborate structures enable the saltatory conduction of action potentials, which is much faster than the pedestrian non-saltatory conduction that occurs along non-myelinated fibers. The picture shows a cross section through a single nerve fiber; the thick, dark zone surrounding the axon is the myelin, which consists of multiple stacked membrane bilayers.97
Cobalamin deficiency causes demyelination or nerve fibers in the central and peripheral nervous system. The neurological, and in advanced stages neuropsychiatric, consequences can be severe. In addition to the disruption of PC and sphingomyelin biosynthesis, deficient methylation of myelin-associated proteins has been considered as another pathogenic mechanism [102].
| 15.4.8 |
Creatine metabolism |
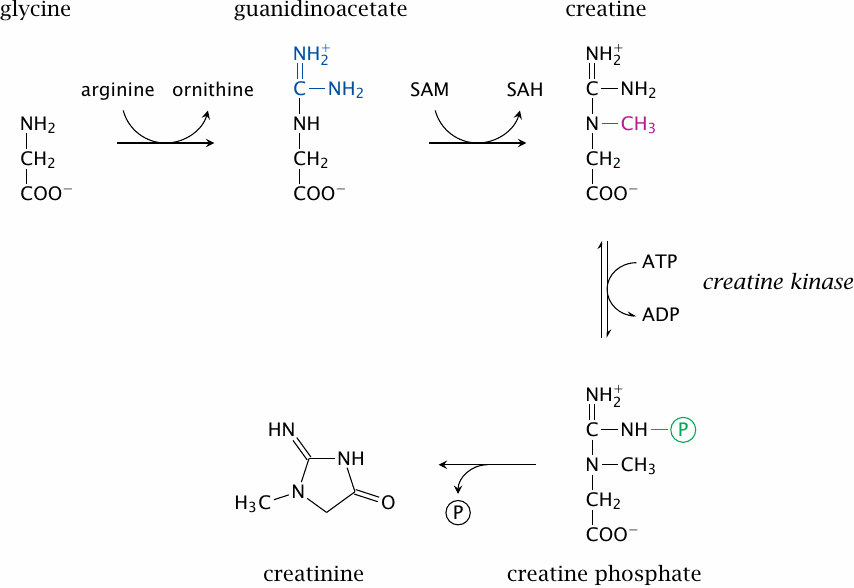
Creatine synthesis involves two steps, which occur in the kidney and the liver, respectively; the second step involves S-adenosylmethionine. Most of the creatine is then accumulated in skeletal muscle. There, it is reversibly phosphorylated to creatine phosphate. This reaction is readily reversible, and the physiological function of creatine phosphate is to form a rapidly available reserve of energy-rich phosphate groups.
Heart and skeletal muscle contain different isoforms of creatine kinase (CK). As with other enzymes, an increased activity of CK in the plasma is a diagnostic indicator of organ damage. High levels of the cardiac isoform of CK are a typical sign of a recent myocardial infarction or of viral myocarditis.
As indicated in the slide, creatine phosphate can undergo spontaneous ring closure to creatine; this reaction consumes approximately 1.5% of the total creatinine pool every day. Creatine is eliminated through the kidneys, predominantly through glomerular filtration. Its concentration in the plasma is used as a diagnostic indicator of kidney function—the lower the glomerular filtration rate, the higher the steady-state concentration of creatinine. In elderly patients, the creatinine level is often used to adjust the dosages of drugs that also undergo renal elimination.
| 15.5 |
Folate and vitamin B12 deficiency syndromes |
Since both folate and B12 are vitamins, deficiency arises from disruptions of intestinal uptake. We therefore need to understand how this intestinal uptake occurs.
| 15.5.1 |
Uptake, transport and storage of folic acid |
- contained in vegetables (Latin folium = leaf)
- synthesized by bacterial flora in the large intestine
- active transport mediates intestinal uptake and renal reuptake, as well as accumulation in the liver
- 50% of all folate is stored in the liver
The high folate content of the liver is consistent with its prominent role in amino acid metabolism.
Since nutrients and vitamins are typically taken up from the small intestine, one might wonder whether or not folate that is synthesized by bacteria in the colon can be taken up by the host. Experiments with instillation of isotopically labeled folate during colonoscopy indicate that uptake from the large intestine is indeed possible [104]. However, the depletion of folate observed in malnutrition or in diseases of the small intestine indicates that colon bacteria do not provide folate in sufficient amounts.
| 15.5.2 |
Causes of folate deficiency |
- malnutrition
- inflammatory bowel diseases
- surgical bowel resection (short intestine syndrome)
- cytochrome P450-inducing drugs
- excessive alcohol consumption—contentious
The first two causes in this list compromise the resorption of folic acid from the small intestine; they apply similarly to many other nutrient deficiency syndromes as well. Alcohol is often cited as a cause of folate deficiency, but no single biochemical mechanism has emerged as the major connection: “Ethanol-associated folate deficiency can develop because of dietary inadequacy, intestinal malabsorption, altered hepatobiliary metabolism, enhanced colonic [that is, bacterial] metabolism, and increased renal excretion” [105].
In studies on the effects of alcohol on folate metabolism with experimental animals, the poor—or lucky, some might say—fellers were usually exposed to excessive dosages, as for example in [106]. In several studies on humans, the correlation between alcohol intake and folate deficiency has been altogether questioned; see for example [107].
Several members of the cytochrome P450 family of enzymes are important in the degradation and inactivation of drugs, which, like folate-dependent metabolism, occurs to a large extent in the liver (see chapter 19). Due to their broad substrate specificities, they cause some modification and inactivation of innocent bystander metabolites also; one of these is folic acid. Various drugs, including ethanol, increase cytochrome P450 activity through transcriptional induction, and long-standing application of such drugs may cause depletion of folic acid through increased metabolism.
| 15.5.3 |
Folate deficiency causes macrocytic anemia |
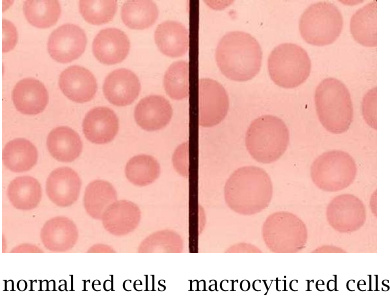
In macrocytic anemia, the number of blood cells is reduced, but the size of the individual cells is increased. With red blood cells, the content of hemoglobin per cell is also increased. On the whole, however, the hemoglobin content of the blood is decreased, which makes this condition a form of anemia.
C1 units derived from THF are required at several points in nucleotide biosynthesis (see slide 15.2.9). The inhibition of nucleotide biosynthesis that results from the lack of THF will interfere with DNA synthesis and cell division more strongly than with protein synthesis. Protein synthesis “outruns” DNA synthesis, leading to the accumulation of more protein per cell between successive cell divisions. In the precursor cells of erythrocytes, this causes a greater than normal hemoglobin content per cell.
Macrocytosis—enlargement of red blood cells—is also often observed in alcoholism, but it may not always be caused by folate deficiency, and the overall content of hemoglobin in the blood may not be reduced.
| 15.5.4 |
The intestinal uptake of vitamin B12 involves multiple carrier proteins |
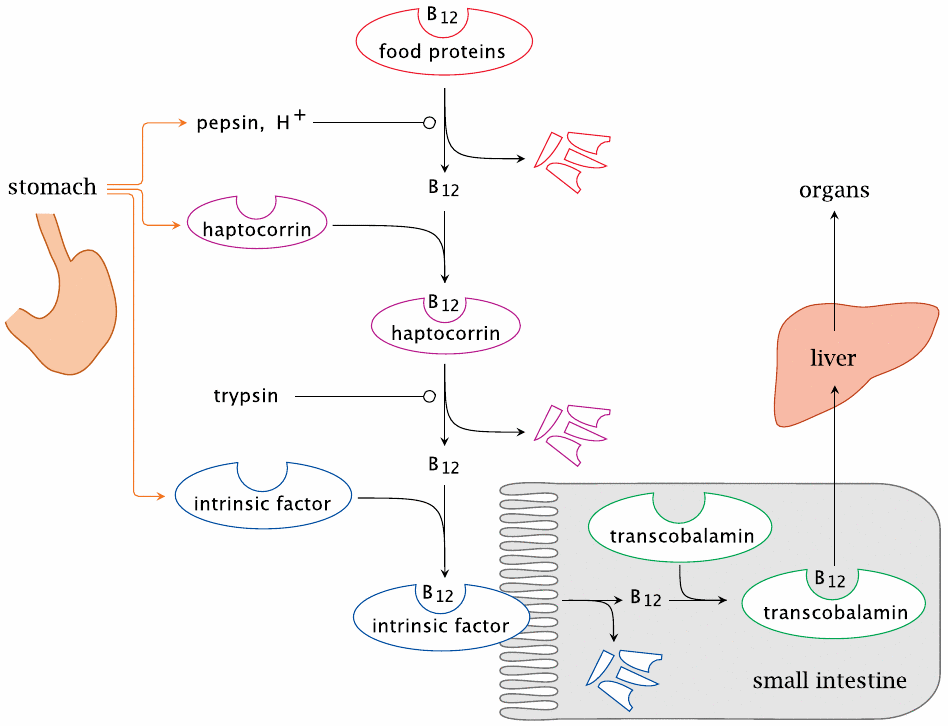
The intestinal uptake of vitamin B12 is a rather intricate affair. The vitamin contained in the food is initially bound to enzymes or other proteins. In the stomach, these food proteins are denatured by gastric acid and cleaved by pepsin. The vitamin thus released is immediately bound by the carrier protein haptocorrin, which is secreted by the gastric mucous membrane.
When the stomach contents reach the small intestine, haptocorrin itself is degraded by trypsin or other pancreatic proteases, and vitamin B12 is released again. The free vitamin is then recaptured by intrinsic factor, a second carrier protein that is also produced by the gastric mucosa (and which up to this point simply came along for the ride). The B12-intrinsic factor complex then travels all the way down to the terminal ileum, the lowermost section of the small intestine, where it is taken up by receptor-mediated endocytosis. Inside the cells of the gut epithelium, vitamin B12 changes carriers for a third time; bound to its final carrier, transcobalamin, the vitamin then reaches the liver and all other tissues beyond.
The daily uptake of vitamin B12 is on the order of only 1-5 µg. The liver stores about 2-5 mg. When uptake is disrupted, this store will last a long while before vitamin deficiency becomes clinically manifest.
| 15.5.5 |
Various causes of B12 deficiency |
| Disease | Pathogenetic mechanism |
| autoimmune gastritis | destruction of the gastric parietal cells that produce gastric acid, haptocorrin, and intrinsic factor |
| pancreatic insufficiency | failure to digest haptocorrin |
| inflammatory bowel disease | disrupted uptake of B12 bound to intrinsic factor |
| receptor deficiencies | disrupted binding and cellular uptake of intrinsic factor or transcobalamin |
Uptake can be disrupted in various circumstances. Crohn’s disease, a major form of inflammatory bowel disease, often most severely affects the terminal ileum, which is where cobalamin, bound to intrinsic factor, must be taken up. In many Crohn’s patients, the terminal ileum is damaged to a point where it needs to be surgically removed.
The classical form of vitamin B12 deficiency, known as pernicious anemia, is caused by an autoimmune disease that destroys the parietal cells, the specific cell type in the gastric mucosa that secretes both haptocorrin and intrinsic factor. The parietal cells also produce gastric acid, the lack of which will compromise the release of B12 from food proteins.
The earliest clinical treatment of pernicious anemia due to vitamin B12 deficiency consisted in the ingestion of copious amounts of homogenized raw liver. Apparently, this was not everybody’s cup of tea, and some patients refused this treatment, in spite of the serious consequences. The situation improved when liver extracts enriched for the vitamin became available. Nowadays, the pure compound is applied orally or intravenously.
As stated above, the amount of cobalamin stored in the liver is many times larger than the daily requirement; the vitamin deficiency therefore tends to become clinically manifest with considerable delay after the disruption of vitamin uptake. This is something to watch out for in patients who have apparently recovered from some intestinal disease or surgery but who have been left with compromised digestive or absorptive function.
Cobalamin has a very high affinity for cyanide, and cyanocobalamin is a common synthetic derivative used as a supplement. Hydroxycobalamin can shed a hydroxide and bind a cyanide ion. In gram amounts, hydroxycobalamin can be used to scavenge free cyanide in acute cyanide poisonings; reportedly, this treatment is superior to the use of oxidative agents that induce the formation of methemoglobin as a cyanide scavenger [108].
| 15.5.6 |
Vitamin B12 deficiency causes disruption of folate-dependent metabolism: the methyl trap ‘hypothesis’ |
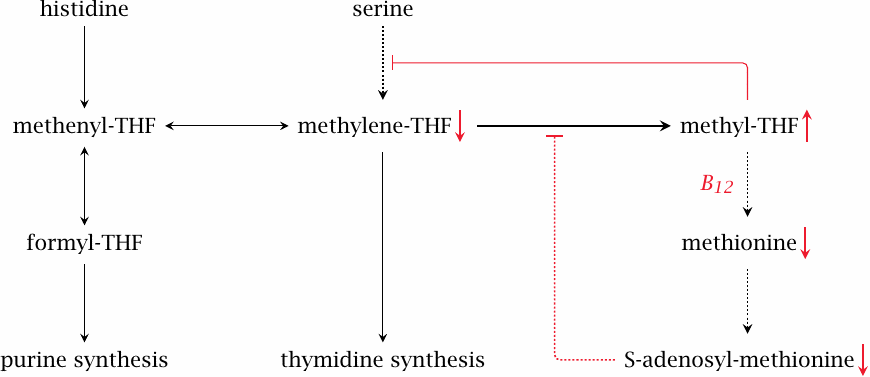
Like folic acid deficiency, a lack of vitamin B12 causes macrocytic anemia, and the changes to the cells found in the peripheral blood and in the bone marrow are indistinguishable between the two conditions. In folate deficiency, the macrocytosis can be explained by the inhibition of nucleotide synthesis (see slide 15.5.3). However, unlike folic acid, cobalamin itself is not required for those synthetic pathways (see slide 15.2.9). How, then, can we account for the clinical resemblance? This is explained by the methyl trap hypothesis.98
When vitamin B12 is deficient, S-adenosylmethionine (SAM) will be lacking (see slide 15.4.2). SAM imposes feedback inhibition on the reduction of N,N’-methylene-tetrahydrofolate to methyl-THF; therefore, the lack of SAM permits excessive accumulation of methyl-THF. The high level of methyl-THF, in turn, inhibits serine hydroxymethyltransferase, which is one of the major reactions that supply C1 units to the THF pool (see slide 15.2.4). The C1-THF pool becomes depleted. In keeping with this explanation, folate substitution does transiently improve the blood count in patients with B12 deficiency.
A lacking capacity for biosynthetic methylation also interferes with the synthesis of phospholipids, which in turn causes demyelination of nerve fibers (see section 15.4). The neurological consequences tend to be more grave in vitamin B12 deficiency than in folate deficiency.