| 17 |
Iron and heme metabolism |
| 17.1 |
Structure and function of heme |
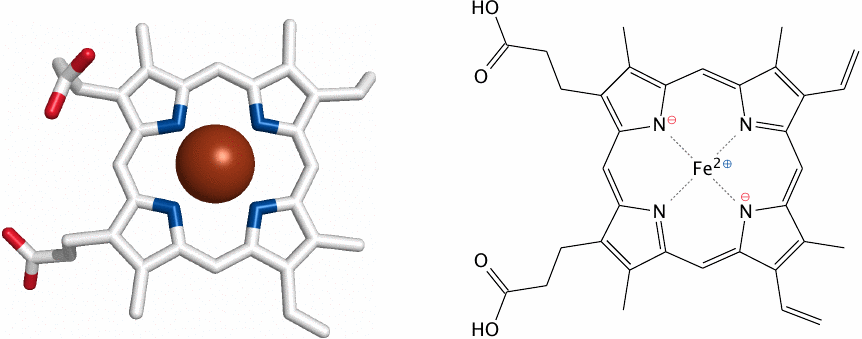
Heme consists of a porphyrin ring that holds a central iron ion. “Porphyr” is the name of a purple-colored gemstone. Heme is colored, too, but its color varies with the protein it is bound to and with the oxidation state of the iron. In addition to hemoglobin, heme is also found in many cytochromes, a term that literally translates as “cell color” and comprises various classes of enzymes and electron carriers. The red-brown colors of tissues like liver, kidney, and brown fat are due to their abundance of heme-containing enzymes or of mitochondria, which contain many heme cofactors within the proteins of the electron transport chain.
In this chapter, we will focus on the biosynthesis and degradation of heme itself. The roles of heme in the respiratory chain and in cytochrome P450 enzymes are covered in chapters 6 and 19, respectively.
| 17.1.1 |
Functions of heme |
Redox chemistry
- electron transport: cytochromes in the respiratory chain
- enzyme catalysis: cytochrome P450, cyclooxygenase, others
Reversible binding of gases
- O2: hemoglobin and myoglobin (80–90% of all heme)
- NO: guanylate cyclase
In hemoglobin and myoglobin, the heme iron remains in the ferrous or Fe2+ state throughout the cycle of oxygen binding and release. In redox-active enzymes and in the respiratory chain, heme regularly goes back and forth between the ferrous and the ferric or Fe3+ states, and sometimes also the ferryl or Fe4+ state.
In cytochrome P450 enzymes, the function of heme is to facilitate the formation of reactive oxygen (see slide 19.2). Similarly, formation of reactive oxygen species (ROS) may also occur as a side reaction wherever heme functions in transport of oxygen or of electrons, and protective mechanisms are required to contain damage by ROS. In hemoglobin, molecular oxygen (O2) may abscond with an extra electron and thereby turn itself into superoxide (O2− •), while the heme iron is left one electron short. Hemoglobin that has lost one electron is called methemoglobin; it is unable to carry oxygen. Its iron is reduced to the ferrous form again by an NADH-dependent enzyme, methemoglobin reductase.102
Protective mechanisms that scavenge reactive oxygen species include the enzymes superoxide dismutase and catalase, as well as antioxidants such as tocopherol (vitamin E; see slide 18.7.11) and ascorbic acid (vitamin C). Organisms that lack such protective mechanisms are anaerobic, that is, they survive only in the absence of oxygen. Anaerobic organisms occur among both prokaryotes and eukaryotes.
| 17.2 |
Heme biosynthesis |
| 17.2.1 |
Overview |
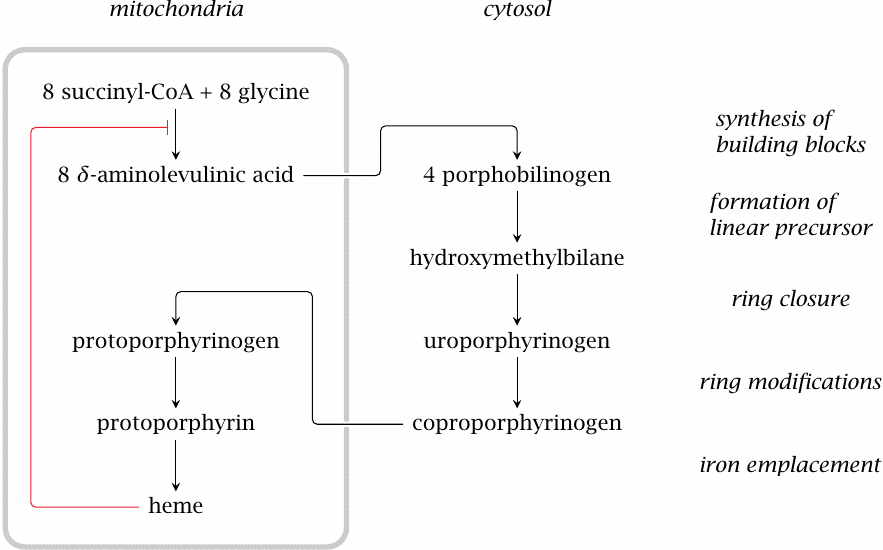
Heme contained in the food is taken up from the intestines, but this is only relevant because of the iron it contains. The organic porphyrin ring is mostly synthesized from scratch. The synthetic pathway is split across two compartments: the initial and final steps occur in the mitochondria, while the intervening ones occur in the cytosol. Heme exerts feedback inhibition on the first step in the pathway.
| 17.2.2 |
The δ-aminolevulinate synthase reaction |
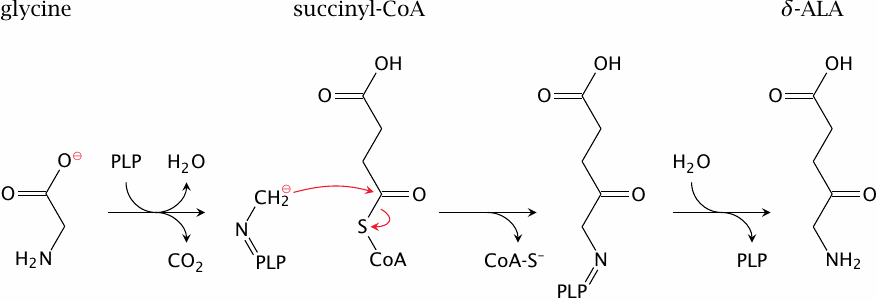
The first reaction in the porphyrin synthesis pathway occurs in the mitochondria. Glycine forms a Schiff base with pyridoxal phosphate (PLP) in the aminolevulinate synthase enzyme. Pyridoxal phosphate acts as a transient “electron sink”, as it usually does and as is illustrated for serine hydroxymethyltransferase in slide 15.2.4. Decarboxylation of the enzyme-bound glycine produces a carbanion intermediate, which reacts with the carbonyl carbon in succinyl-CoA.
The aminolevulinate synthase reaction is the committed step of heme synthesis and is subject to feedback inhibition by heme, the final product of the pathway. The reaction product, δ-aminolevulinic acid (δ-ALA), is transported to the cytosol, where the next four reactions occur.
| 17.2.3 |
The porphobilinogen synthase reaction |
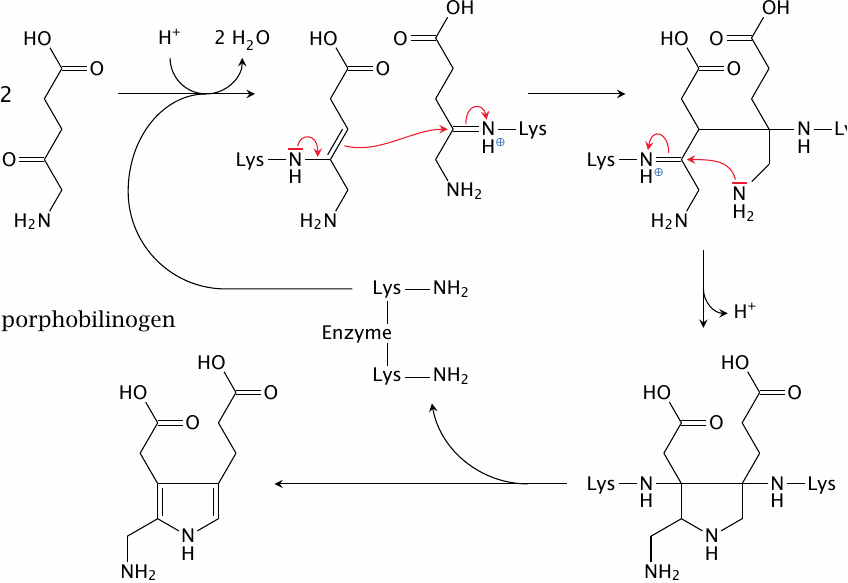
In the second reaction of heme synthesis, porphobilinogen synthase condenses two molecules of δ-ALA into one molecule of porphobilinogen, with the elimination of two molecules of water. At the outset, both substrate molecules form Schiff bases with two separate lysine residues in the active site. The water is already lost at this stage; however, the more interesting reaction steps that lead to the formation of the pyrrole ring still lie ahead. It should be noted that the reaction mechanism depicted here is still somewhat tentative [133].
| 17.2.4 |
A substrate analogue next to zinc inside the active site of porphobilinogen synthase |
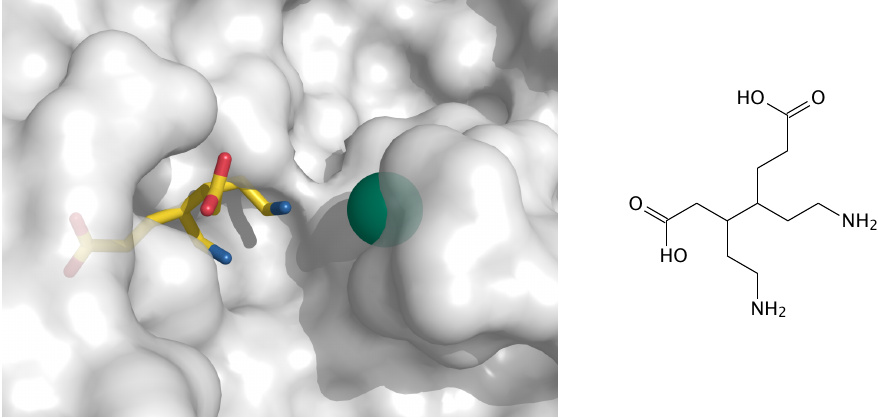
This pretty picture (rendered from 1pv8.pdb) shows a model compound (structure on the right) inside the active site of porphobilinogen synthase, next to a zinc ion. It sheds little light on the reaction mechanism, or on the specific role of zinc. The only point I want to make here is that zinc is important for this enzyme. In lead intoxication, the zinc in porphobilinogen synthase is dislodged by lead, which inhibits the enzyme and therefore the biosynthesis of heme and hemoglobin.
| 17.2.5 |
Porphobilinogen deaminase synthesizes hydroxymethylbilane |
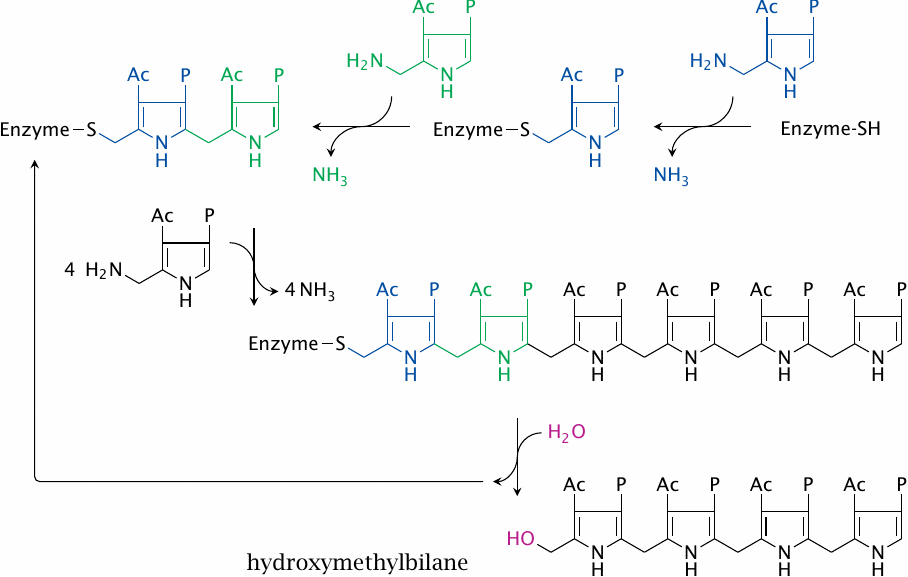
Porphobilinogen deaminase (or hydroxymethylbilane synthase) turns four molecules of porphobilinogen into one linear tetramer. The process involves the priming of the enzyme with two porphobilinogen subunits (shown in blue and green) which remain with the enzyme molecule throughout and serve as a prosthetic group. Another unusual feature of this enzyme is that it uses ammonia as a leaving group. (In this scheme, Ac and P denote the acetate and propionate groups that are part of the porphobilinogen molecule; compare slide 17.2.3.)
| 17.2.6 |
Synthesis of uro- and coproporphyrinogen III |
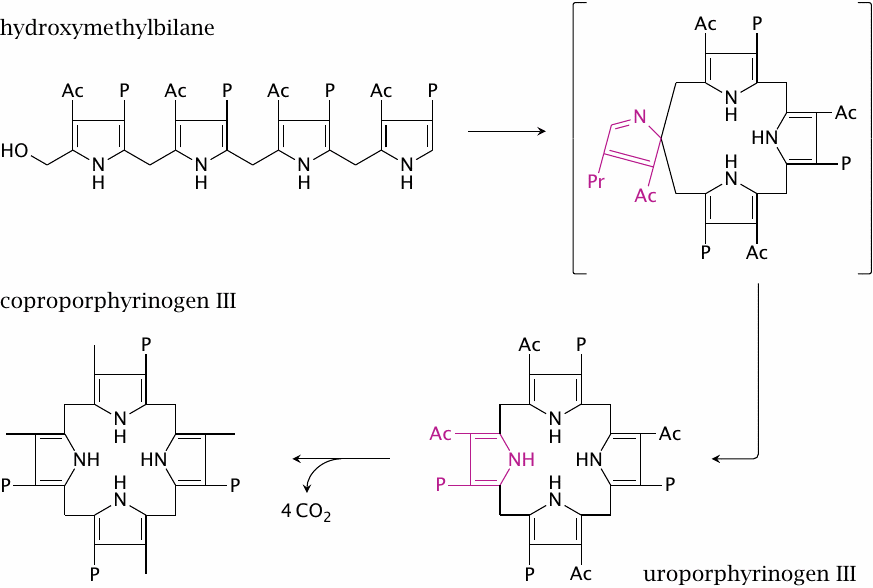
Hydroxymethylbilane, when left alone, cyclizes spontaneously, giving rise to a ring with fourfold rotational symmetry, with acetyl groups and propionyl groups alternating regularly around the perimeter of the molecule. The product of this spontaneous reaction is named uroporphyrinogen I.
Nature, however, prefers another isomer, namely, uroporphyrinogen III. To forestall its spontaneous cyclization into uroporphyrinogen I, hydroxymethylbilane is captured and processed directly by uroporphyrinogen III cosynthase, which forms a complex with porphyrinogen deaminase. The cosynthase reaction proceeds via the improbable-looking spiro intermediate shown here within brackets, which leads to the inverted orientation of the acetate and propionate side chains on the fourth ring in uroporphyrinogen III.
The next enzyme, uroporphyrinogen III decarboxylase, decarboxylates all four acetate groups to methyl groups. This gives coproporphyrinogen III, which again enters the mitochondria.
| 17.2.7 |
Final steps in heme synthesis |
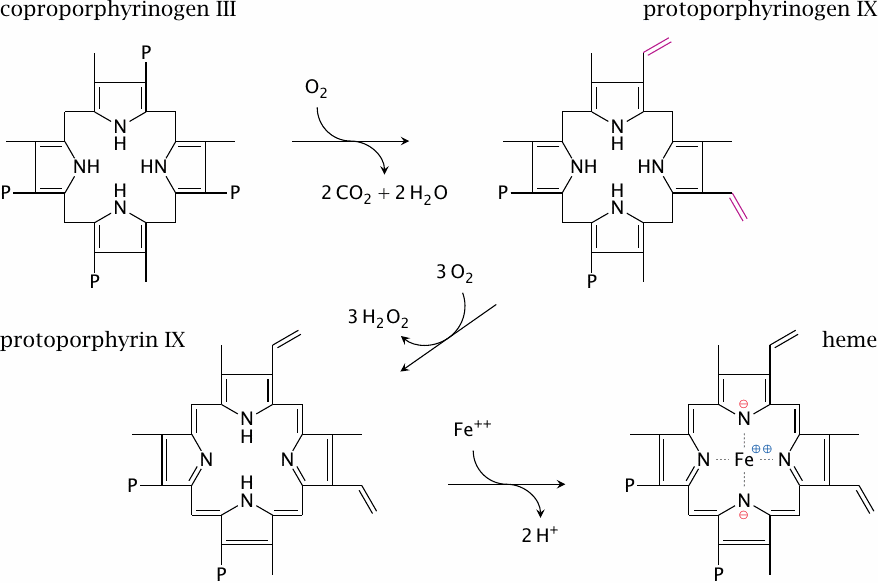
Oxidative decarboxylation converts two of the four propionate residues to vinyl groups, which produces protoporphyrinogen IX. Dehydrogenation to protoporphyrin IX is the final ring modification. Ferrochelatase then inserts the iron, which completes heme synthesis.
While the ferrochelatase reaction looks rather trivial and indeed can proceed with appreciable rate spontaneously, the enzyme is important: when it is absent, protoporphyrin IX accumulates, which gives rise to a clinically manifest form of porphyria [134].
Heme retains the vinyl groups that were introduced at the stage of protoporphyrinogen IX. Addition of the sulfhydryl groups of cysteine side chains across these vinyl groups can produce covalent bonds between heme and some of its apoproteins. Such covalent attachment occurs for example in cytochrome C oxidase, but it does not happen in hemoglobin or myoglobin.
| 17.3 |
Disruptions of heme synthesis |
- iron depletion
- hereditary enzyme defects (porphyrias)
- vitamin B6 deficiency—inhibition of aminolevulinate synthase
- lead poisoning—inhibition of porphobilinogen synthase
Hereditary deficiencies are known for each of the enzymes in the synthetic pathway. Clinical symptoms are due to both lack of heme and to the accumulation of biosynthetic intermediates. Heme exercises feedback inhibition on the first enzyme in the synthetic pathway, that is, δ-ALA synthase. A lack of heme therefore disinhibits this enzyme and amplifies the accumulation of intermediates upstream of the enzyme defect in question. Backed-up synthetic intermediates often undergo spontaneous conversion to aberrant products.
Inhibition of heme synthesis can also result from causes other than enzyme defects. The most common cause is iron depletion; another one is deficiency of vitamin B6, which can result from malnutrition or, in inflammatory intestinal diseases, from malabsorption. Vitamin B6 (pyridoxin) is the precursor of pyridoxal phosphate, the coenzyme in aminolevulinate synthase. Lead intoxication causes inhibition of porphobilinogen synthase (see slide 17.2.3); it is now less common than it used to be.
| 17.3.1 |
Disruption of heme synthesis causes microcytic, hypochromic anemia |
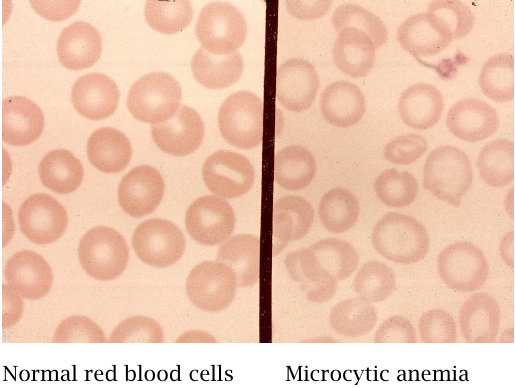
Regardless of the underlying cause, the inhibition of heme synthesis will result in red blood cells that are smaller and contain less hemoglobin than normal ones. This condition is named microcytic, hypochromic anemia. The term “microcytic” refers to the reduced cell size, whereas “hypochromic” denotes the reduced intracellular hemoglobin concentration.
| 17.3.2 |
Porphyria cutanea tarda (PCT) is caused by uroporphyrinogen decarboxylase deficiency |
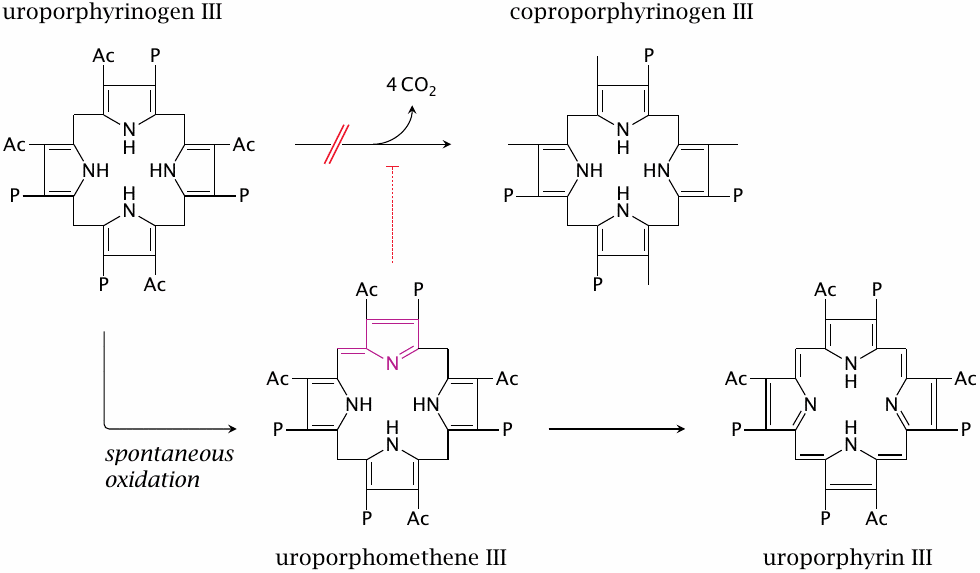
The accumulation of porphyrin precursors in various enzyme defects in porphyrin synthesis can cause photosensitization of the skin. The clinical picture can vary a bit, depending on the specific intermediate. As an example, we will consider the disease porphyria cutanea tarda (translated: chronic porphyria of the skin), which is the most common form of porphyria. Here, the deficient enzyme is uroporphyrinogen III decarboxylase (see slide 17.2.6). This leads to the accumulation of an aberrant metabolite, uroporphyrinogen III. Its likewise aberrant precursor uroporphomethene III is an inhibitor of the already deficient decarboxylase, which will tend to make things worse.
| 17.3.3 |
Laboratory and clinical findings in PCT |
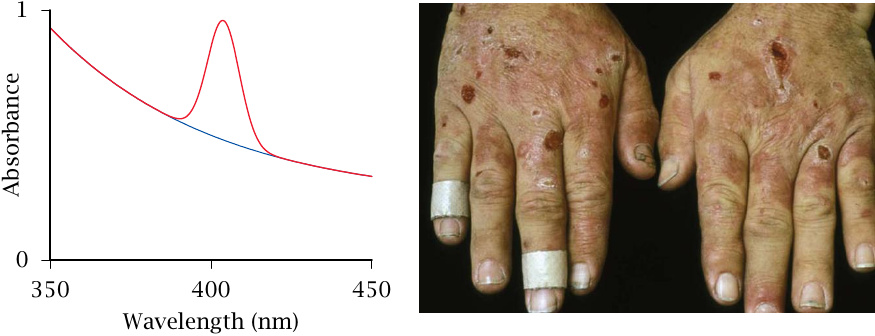
The accumulating uroporphyrinogen distributes throughout the body and becomes oxidized non-enzymatically to the non-physiological product uroporphyrin III. In the skin, uroporphyrin III can absorb photons and then react with molecular oxygen to produce reactive oxygen species; the latter inflict the skin tissue damage that is illustrated in the slide. An important aspect of treatment is the protection of skin from direct sunlight.
The absorbed wavelength range (or absorption spectrum) differs between the various porphyrins. Uroporphyrin III has an absorption peak at 405 nm, which is at the blue end of the visible spectrum; this peak is readily detectable in blood serum samples (left). The sun light is more intense in the visible range than in the UV range. Sun screen lotion, which is designed to absorb UV light but not visible light, will not prevent photosensitization by uroporphyrin III.
| 17.3.4 |
Causation of porphyria cutanea tarda |
- hereditary—rare, autosomal dominant; enzyme defect is manifest in all tissues
-
sporadic—exogenous, or related to a genetic defect in iron uptake regulation
- caused by alcohol, halogenated hydrocarbons, other toxic substances
- enzyme activity lacking in the liver but not erythrocytes and other tissues—enzyme is functional but inhibited by interfering metabolites
- iron overload seems important in both hereditary and sporadic forms
Sporadic PCT is often associated with disturbances of iron homeostasis. Approximately two thirds of sporadic PCT patients carry a deficient allele for the iron regulator HFE. In homozygous form, this gene defect causes hemochromatosis, a disease that is characterized by severe iron overload. HFE knockout mice show increased intestinal expression and activity of iron uptake transporters. Excess iron may facilitate the non-enzymatic oxidation of uroporphyrinogen to uroporphyrin. An intermediate of this oxidation, uroporphomethene (see slide 17.3.2), inhibits uroporphyrin decarboxylase, which would account for the role of iron overload in pathogenesis [135].
Iron overload of the liver can also occur in chronic infections and in other chronic inflammatory diseases (see section 17.5). Blood letting—which depletes iron—is reportedly beneficial in PCT, regardless of the cause of the iron overload.
| 17.3.5 |
A defect of ferrochelatase causes erythropoietic protoporphyria |
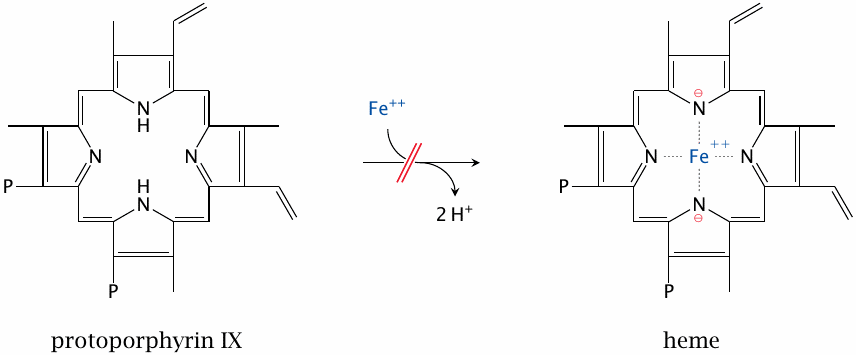
While the final reaction in heme synthesis—namely, the emplacement of Fe++ into the completed protoporphyrin ring—looks like a facile reaction, and indeed proceeds with appreciable speed without ferrochelatase, the enzyme is still needed to make the reaction go fast enough. If the enzyme is deficient, protoporphyrin accumulates and causes skin manifestations similar to those observed in porphyria cutanea tarda.
| 17.3.6 |
Acute intermittent porphyria (AIP) |
- deficiency of porphobilinogen deaminase, autosomal dominant
- excessive synthesis of δ-ALA in liver
- surplus porphobilinogen in urine—urine is colored red
-
δ-ALA inhibits the GABAA receptor, causing
- psychiatric symptoms (‘organic psychosis’)—too often misdiagnosed and mistreated
- abdominal pain (neuropathic)
- episodes can be induced by drugs
The genetic defect in acute intermittent porphyria concerns the enzyme porphobilinogen deaminase. Both porphobilinogen and δ-ALA accumulate. Porphobilinogen is excreted with the urine and, through spontaneous oxidation, forms a characteristic red pigment. The more severe clinical symptoms, however, are due to accumulation of δ-aminolevulinate. This metabolite competitively inhibits the binding of γ-aminobutyrate (GABA), an inhibitory neurotransmitter, to its receptors [136], which likely causes both the psychiatric symptoms (agitation, confusion) and the neurological ones (nausea, abdominal pain) in AIP.
The dysregulation mainly affects heme synthesis in the liver. As the name of the disease indicates, it is not always manifest but only intermittently. Heme is the prosthetic group of cytochrome P450 enzymes, which are important in drug metabolism and are induced in the liver by various drugs (see slide 19.2.2). It appears that ALA synthase is induced along with the cytochrome P450 enzymes, and AIP attacks are often triggered or aggravated by the application of such drugs.
Specific drugs that induce cytochrome P450 and ALA synthase include barbituric acid derivatives and carbamazepine, which were, and occasionally still are, used in the treatment of psychiatric symptoms. Fatal outcomes have occurred when AIP patients were misdiagnosed and treated with barbituric acid derivatives.103 One element of the correct therapy consists in the application of heme, which inhibits ALA synthase.
| 17.4 |
Heme degradation |
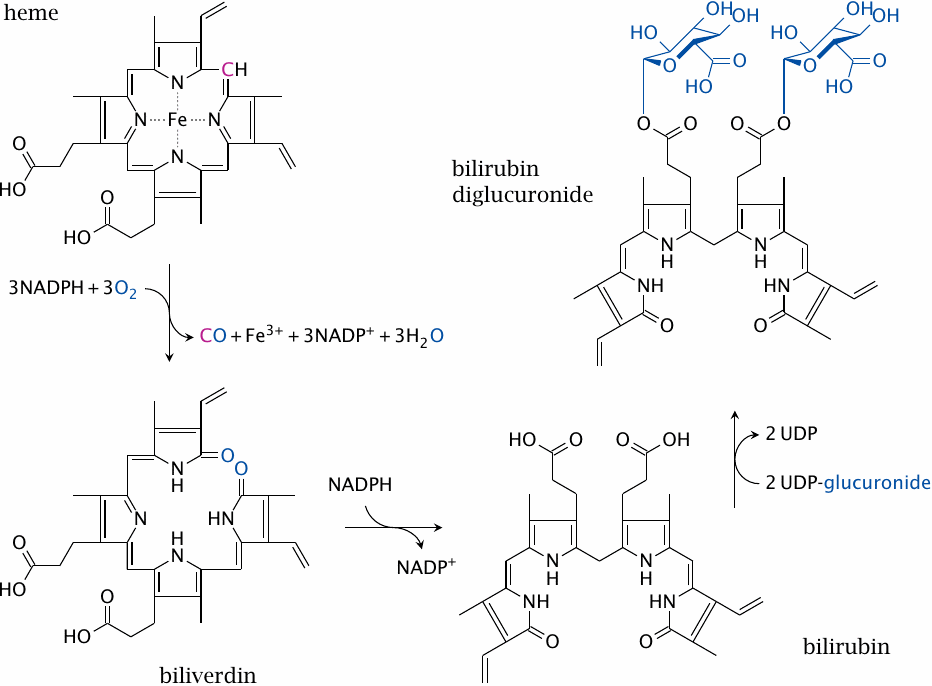
Red blood cells have a regular lifespan of 120 days (although it can be considerably shorter in some diseases). At the end of this lifespan, they are captured and ingested by phagocytes in the spleen and the liver. When the globin protein is proteolytically degraded, heme is released. Heme itself undergoes degradation mostly in the liver. Ring cleavage by heme oxygenase produces biliverdin, which is in turn reduced to bilirubin. Some bilirubin is excreted into the bile as such; however, the greater share is first conjugated with glucuronic acid by UDP-glucuronosyltransferase (form 1A1) and excreted thereafter. The major transport protein responsible for excretion of the diglucuronide is an ABC transporter (ABCC2), the same one that also secretes bile acids (see slide 11.5.3).
In the large intestine, part of the conjugated bilirubin undergoes deconjugation by bacterial β-glucuronidases. In the anaerobic environment that prevails inside the colon, the released bilirubin subsequently undergoes reduction, again by bacterial enzymes, to variously colored pigments that produce the stool color. Another reduction product, urobilinogen, is taken up and excreted with the urine, causing the yellow color of the latter.
| 17.4.1 |
Jaundice |
Accumulation of bilirubin in the body. Causes:
- increased production: hemolytic anemia (premature decay of red blood cells)
- decreased conjugation: enzyme defect, liver disease
- decreased excretion of conjugated heme: deficiency of ABCC2 transporter (Dubin-Johnson syndrome)
- mechanically blocked excretion: bile duct blocked by bile stone or tumor
Some fairly simple clues can narrow down the cause of jaundice in a given patient. If excretion of bilirubin is blocked, the pigments derived from it will be absent, and the stool will have a grayish color.
Hemolysis consists in the accelerated decay of red blood cells; it may result from biochemical causes such as glucose-6-phosphate dehydrogenase deficiency (see slide 9.4), or from immunological ones such as Rhesus incompatibility. In hemolysis, the serum level of unconjugated bilirubin will be more strongly increased than that of the diglucuronide. On the other hand, when the flow of the bile is backed up, the conjugated bilirubin will spill back into the serum and will be increased.
Liver diseases such as hepatitis can affect synthesis, conjugation and biliary secretion of bilirubin to various degrees, and either form of bilirubin can be more strongly increased than the other.
| 17.4.2 |
Enzyme defects in bilirubin conjugation by UDP-glucuronosyltransferase |
- transient, usually mild: neonatal jaundice
- genetic, mild: Gilbert syndrome—asymptomatic jaundice
- genetic, severe, rare: Crigler-Najjar syndrome
Neonatal jaundice is a normal event that is caused by a transiently low level of UDP-glucuronosyltransferase 1A1.104 In most cases, it does not cause any problems and simply goes away within two weeks after birth, as the level of enzyme activity increases. If the serum level of bilirubin gets too high, however, it may accumulate in the brain and cause neurological problems (see next slide). To prevent this, newborns can be treated with phototherapy (see slide 17.4.4).
Crigler-Najjar syndrome is due to the genetic deficiency of UDP-glucuronosyltransferase 1A1. It initially becomes manifest as neonatal jaundice; however, since the gene defect is permanent, the situation doesn’t improve with time. As in many other gene defects, there are variants with total or partial disruption of enzyme activity. Milder cases are classified as Gilbert’s syndrome; the boundary between Gilbert’s and Crigler-Najjar syndrome will be somewhat arbitrary. When residual enzyme activity is present, it may be increased with drugs such as phenobarbital that transcriptionally induce it.
As in neonatal jaundice, phototherapy is also used in Crigler-Najjar syndrome, but its efficiency decreases with time, since growth reduces the body’s surface-to-volume ratio, and therefore a diminishing fraction of the bilirubin in the body can be reached by the illumination. The disease is best treated with liver transplants, as the transplanted liver will not be affected by the underlying gene defect and be able to conjugate and excrete bilirubin.
| 17.4.3 |
Bilirubin encephalopathy (“kernicterus”) |
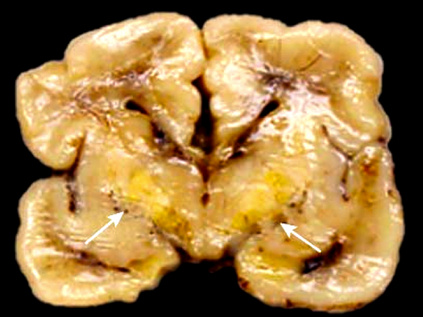
This slide shows a brain section from a newborn with severe bilirubin encephalopathy. The yellow color in the deeper structures of the forebrain, the so-called basal ganglia, is due to bilirubin accumulation. Through an as yet unknown biochemical mechanism [137–139], bilirubin causes damage to the basal ganglia, which results in motor dysfunction and other neurological symptoms.
| 17.4.4 |
Photoisomerization products of bilirubin |
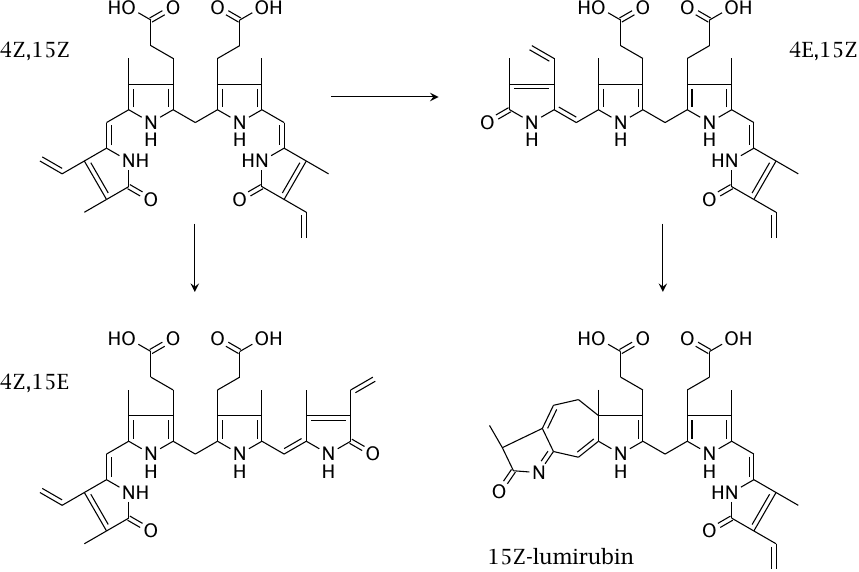
In phototherapy, bilirubin absorbs photons and subsequently undergoes cis-trans isomerization across the two remaining double bonds between the pyrrole rings of the bilirubin molecule, as well as ring formation [140]. This slide shows some of the photochemical reaction products. The 4Z,15Z isomer of bilirubin (top left) is the one that is produced directly by biliverdin reductase, and which is eliminated very slowly in the unconjugated form. The other isomers are eliminated more rapidly; the fastest rate of elimination is observed with lumirubin (cyclobilirubin).
While the absorption maximum of bilirubin is in the blue wavelength band, green light reportedly produces lumirubin more efficiently, and it also induces less cytotoxic byproducts in cell culture models [141]. It seems, however, that blue lamps are still more widely used in practice, and the literature makes no mention of significant side effects of blue light, even in the long-term treatment of Crigler-Najjar patients [142].
| 17.4.5 |
Sn-mesoporphyrin, an inhibitor of heme oxygenase |
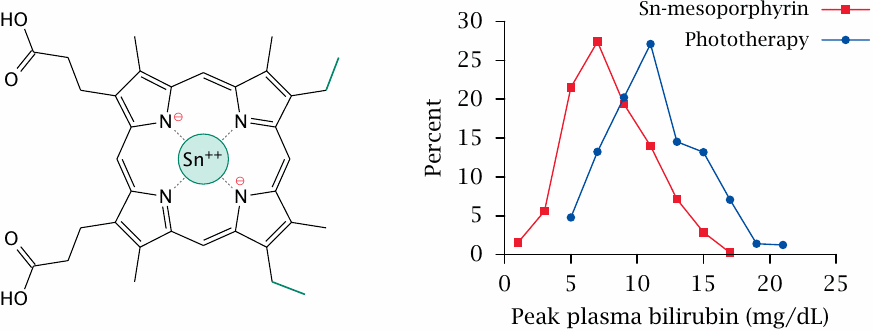
The inhibition of heme oxygenase with Sn-mesoporphyrin has been used successfully in clinical studies to treat neonatal jaundice. The study summarized in this slide examined the effectiveness of Sn-mesoporphyrin in the treatment of newborns with glucose-6-phosphate dehydrogenase deficiency (see slide 9.4). In this condition, the lifespan of red blood cells is diminished, which increases the rate of heme degradation; newborns therefore are at an increased risk of severe jaundice.
Remarkably, a single injection of the drug was sufficient to reduce the peak levels of bilirubin to a greater extent than the reference treatment (phototherapy). It should be noted that both forms of treatment sufficed to keep the peak bilirubin levels below 25 mg/dL, which is considered sufficient to avoid damage to the brain. Phototherapy currently remains the standard treatment in clinical practice. Figure prepared from original data in [143].
| 17.4.6 |
Is CO a signaling molecule, like NO? |
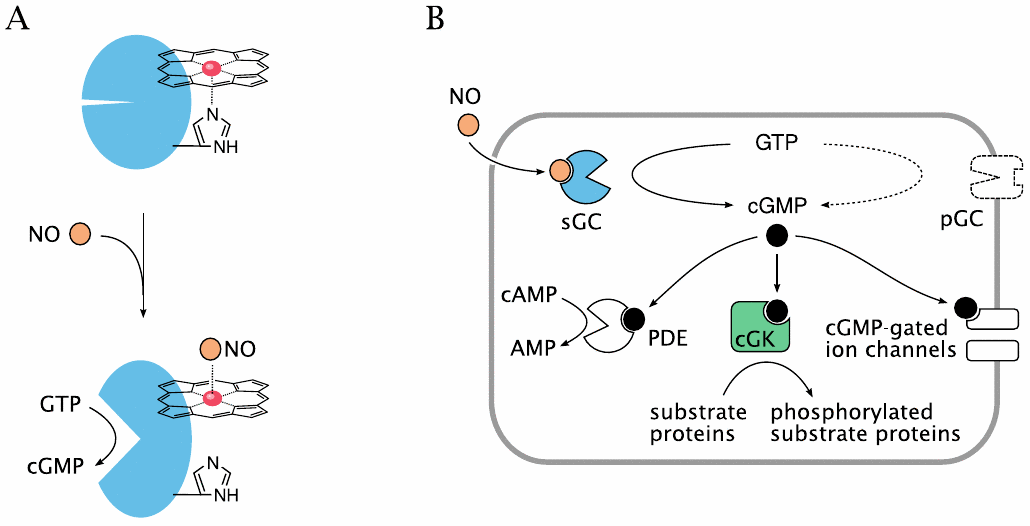
Nitric oxide is an important signaling molecule. As a small molecule, it can easily diffuse out of one cell and into another. Inside the target cell, it binds to soluble guanylate cyclase (sGC). Interestingly, the NO-binding site on sGC is a heme molecule. Binding of NO to one face of the heme releases a histidine side chain on the other, which causes a conformational change and activation of the sGC molecule.
In vitro experiments show that CO can also bind and activate sGC. It has therefore been proposed that heme oxygenase, which produces CO, has a regulatory role in addition to its metabolic one. However, while this idea has been around for awhile, I have not come across solid evidence that supports a significant signaling role of CO in vivo.
| 17.5 |
Iron uptake, transport and storage |
- uptake in the small intestine: Fe2+—free or bound to heme
- transient storage as ferritin inside the intestinal epithelia
- transport in the blood: Fe3+—bound to transferrin with very high affinity
- cellular uptake: endocytosis of transferrin, release of iron in acidic endosome
- storage: intracellular ferritin particles
- depletion: scaled-off cells, blood loss, breast milk
Only ferrous iron (Fe2+) can be taken up from the small intestine, so that is what we give to patients. Still, during passage through the intestine, much of the ferrous iron is oxidized to the ferric form (Fe3+), which means that intestinal uptake is not very efficient.
The very high affinity of transferrin for iron means that there is practically no free iron in the blood serum. Bacteria, like human cells, require iron for growth; therefore, keeping free iron very low is an important non-specific immune mechanism. Many pathogenic bacteria produce their own high-affinity iron-binding molecules (siderophores) to acquire iron within this iron-depleted environment.
In chronic infections, free iron in the serum is reduced even below the normal low value, presumably in an attempt by the immune system to starve microbial pathogens of iron. The same also happens in tumor patients and non-infectious inflammatory diseases such as rheumatism, since the immune system is not smart enough to tell the difference between these and infections. As a result, an anemia develops in which the blood cells look just like the ones in true iron depletion. However, in contrast to the latter, the level of cellular storage iron will be increased in this case—iron is not lacking, it is just being kept out of circulation. If such iron sequestration is observed in a patient, one must search for the underlying disease that causes it.
Loss of iron occurs with cells being scaled off from the skin and intestinal epithelia, with blood loss (menstruation, blood donations), with diaplacental transfer to a growing fetus, and with breast milk. Mothers who have many children within a relatively short period of time have a good chance to incur iron depletion, so this is something to watch out for as a family physician.
| 17.5.1 |
Structure of ferritin |
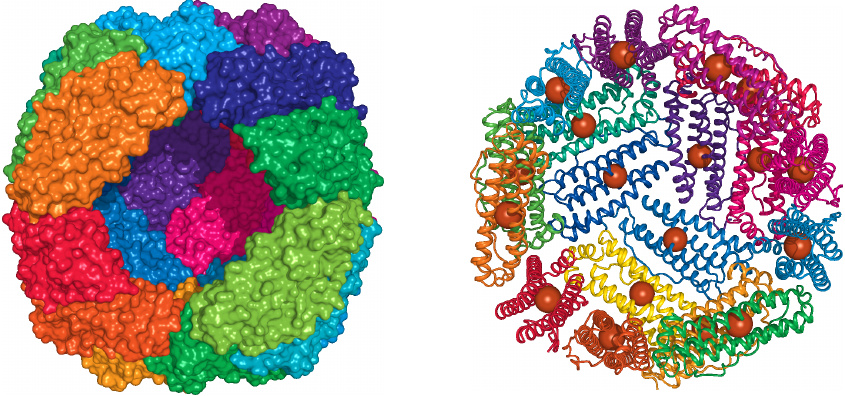
Ferritin is a hollow protein particle that consists of 24 identical subunits. Up to ~4,500 iron ions can be stored inside the particle, in the form of FeOOH.
On the left, two neighboring subunits have been removed from the front of the sphere, and we are peeking into the interior of the shell, which in vivo would be packed with iron. On the right, some more subunits have been removed, and the iron ions actually contained in the crystal structure—just one per subunit—are shown as spheres. Can you figure out why the crystallographers did not try to obtain a structure of the fully loaded particle? Rendered from 1fha.pdb.
| 17.5.2 |
Ferritin in the small intestine regulates iron uptake |
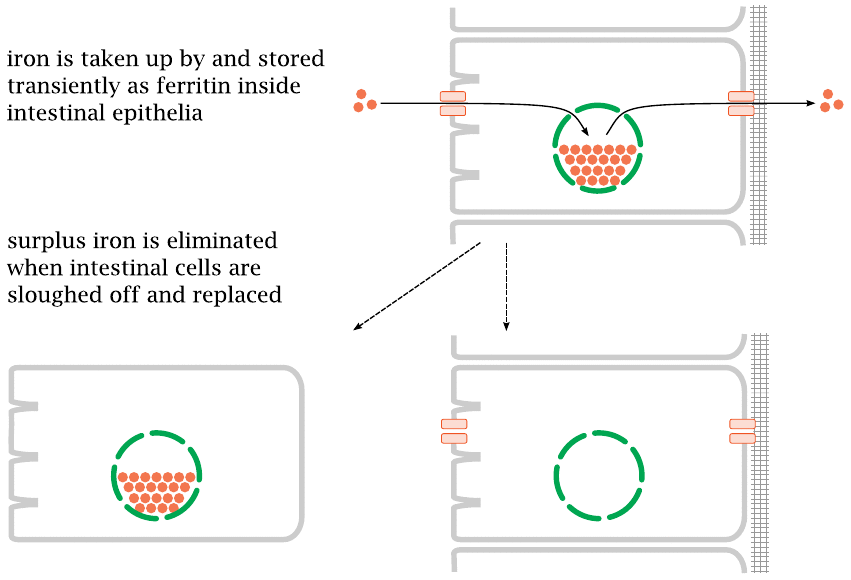
Much of the iron regulation is accomplished right in the small intestine. Iron that is taken up is initially stored inside the epithelial cells; surplus iron that is not requested from this store is simply lost when the epithelial cells are replaced, which happens about once a week.
| 17.5.3 |
Hemosiderin in liver tissue |
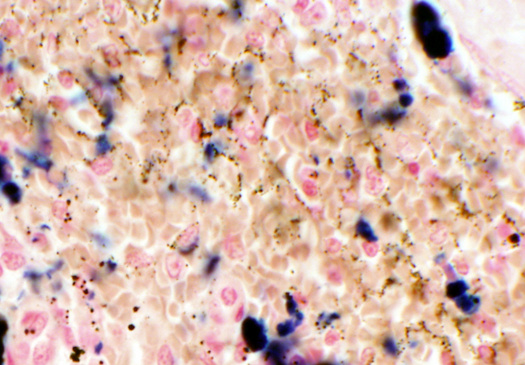
In diseases that cause iron overload, the storage capacity of ferritin will be exceeded. Excess iron will then precipitate around the ferritin particles and cause them to aggregate. These iron-rich aggregates are called hemosiderin.
Iron overload can occur as a result of multiple blood transfusions, or of genetic defects that interfere with iron transport. An example is a defect of ceruloplasmin, a copper-containing serum protein that oxidizes Fe2+ to Fe3+.
In the figure, the brown stipples represent the hemosiderin. The dark blue blotches are out of focus and are just precipitated dye particles, that is, artifacts; this tends to happen if dye solutions stand around on the shelf for too long.