| 16 |
Nucleotide metabolism |
| 16.1 |
Introduction |
| 16.1.1 |
Functions of nucleotides in biochemistry |
- Building blocks of nucleic acids
- Cosubstrates and coenzymes
- Signaling
I’ll be wildly optimistic and assume that you remember a thing or two about nucleic acids, so we will mostly skip this topic.
Nucleotides are very important as cosubstrates in metabolism. As you know, ATP occurs everywhere, but GTP, CTP, and UTP drive some biochemical reactions as well.
Adenosine and adenine nucleotides also function as signaling molecules; for example, ADP is important in thrombocyte activation. Caffeine is an antagonist at adenosine receptors, which tells us that adenosine is important in the regulation of vigilance. Cyclic AMP (slide 7.5.4)and cyclic GMP (slide 9.3.6) function as intracellular second messengers.
| 16.1.2 |
Structures of PAPS, acetyl-CoA, and NAD |
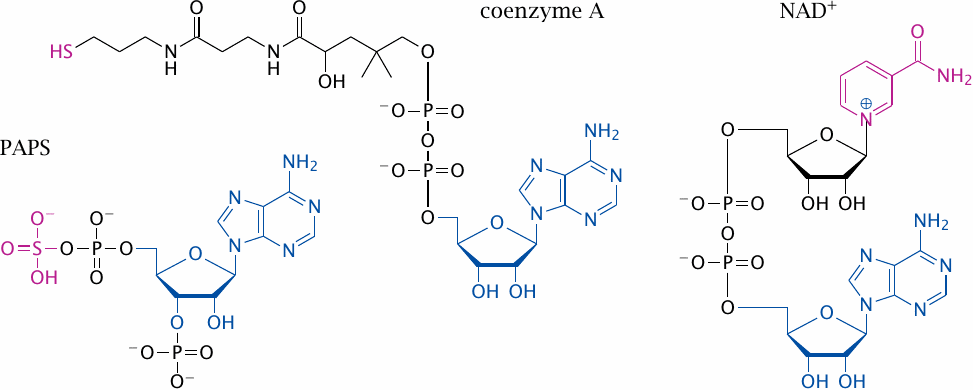
Nucleotides also occur as parts of more complex cosubstrates and coenzymes, three of which are shown here. These three molecules have very different “business ends.” In coenzyme A, the business end is the thiol group that becomes bound to the substrate, and in NAD+ it is the nicotinamide moiety that undergoes reversible reduction and oxidation. In 3′-phosphoadenosine-5′-phosphosulfate (PAPS), the key feature is the mixed phosphosulfate anhydride that activates the sulfate toward transfer, in much the same way as the terminal phosphate is activated in ATP. In methylcobalamin, the chemically active center is the cobalt ion that facilitates carbon bond cleavage and formation (see section 15.4).
The adenosine moiety found in each of these molecules, shown in blue, does not directly participate in any of the catalytic functions; instead, it just serves as a tag to facilitate recognition of the cosubstrates by the corresponding enzymes. Why, then, do cosubstrates so often possess nucleotides as their binding tags, rather than for example amino acids or peptides? A plausible answer is provided by the RNA world hypothesis.
| 16.1.3 |
The RNA world hypothesis |
The RNA world is a hypothetical early stage of evolution. In this early world, RNA was the predominant macromolecule. RNA not only stored and propagated genetic information, a role that is almost universally filled by DNA in current life forms, but it also realized and expressed this information, which in current life forms is mostly accomplished by proteins.
The empirical basis of the RNA world hypothesis is that RNA can indeed assume the role of DNA, as it still does in RNA viruses and viroids, and that it can also have catalytic activity, as is the case in ribosomes and smaller ribozymes.
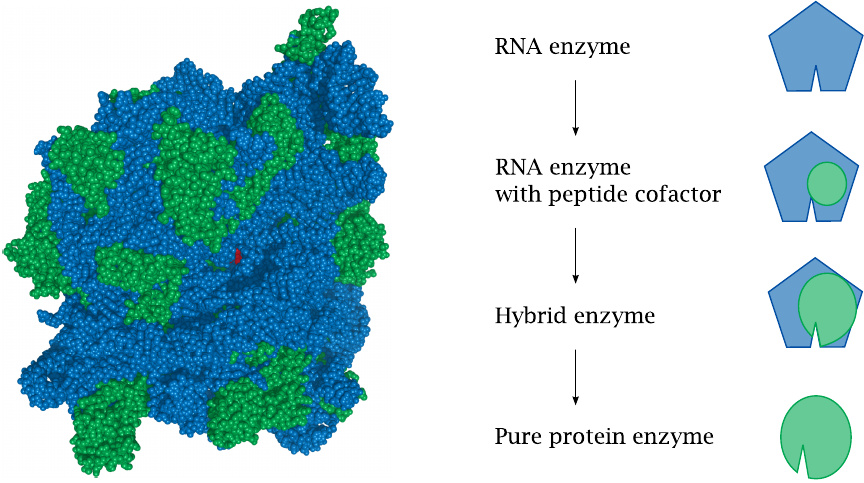
The structure of the ribosome also suggests an evolutionary path along which RNA enzymes may have been replaced by protein enzymes in evolution. The catalytic centers of a ribosome (left) consist entirely of RNA (blue). The tiny red dot in the center represents the antibiotic chloramphenicol, lodged in one of the active sites of a bacterial ribosome; we are peeping at it through the ribosome’s peptide exit tunnel. Ribosomal proteins (green) serve in structural and auxiliary roles.
Like ribosomes, other RNA enzymes may at first have coopted peptides as structural components, and possibly as coenzymes. The peptides may then have grown into ever more important roles within the hybrid molecules, until they took over entirely and made the RNA component obsolete.
| 16.1.4 |
Why have cosubstrates become fossilized, whereas enzymes have not? |
In a world dominated by RNA, one would expect cosubstrates and carrier molecules to contain some nucleotide moieties also. However, if the above scenario for the evolutionary replacement of RNA enzymes by protein enzymes is correct, why did a similar replacement not occur with cosubstrates?
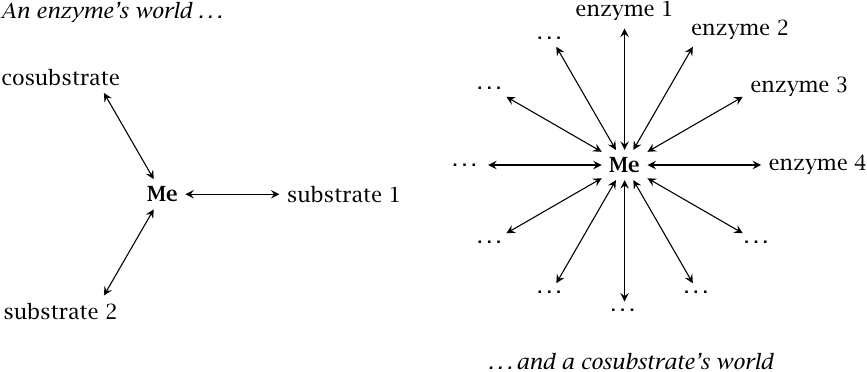
Let us consider the world from the perspective of an enzyme and of a cosubstrate, respectively. An enzyme’s world is simple, containing one or a few substrates and maybe a cosubstrate or coenzyme. If we make some random change to the structure of the enzyme, there is a reasonable chance that the interactions with all of these ligands will remain intact.
In contrast, a cosubstrate such as ATP or NADH inhabits a much more complex world, containing a very large number of interacting enzymes. Chances are that a change to its molecular structure would disrupt its interaction with some enzyme molecule or other that is essential to the survival of the cell. Structural changes to cosubstrates are therefore “forbidden,” and they may well look now exactly as they did shortly after life first arose billions of years ago.99
| 16.2 |
Overview of metabolic pathways for nucleotides |
| 16.2.1 |
Metabolic routes and pathways of nucleotides |
- De novo synthesis
- Intestinal uptake of nucleosides
- Endogenous turnover (partial degradation/salvage)
- Degradation and excretion
Synthesis of nucleotides from scratch occurs in all tissues, and its capacity is sufficient to fully cover the needs of the organism; we do not require any nucleotides or bases in the diet. Nevertheless, nucleic acids contained in the diet are broken down to nucleosides, which are taken up with high efficiency and degraded (see section 16.4).
The pathways for the degradation of endogenous nucleotides have some overlap with those for dietary ones. Intermediates of nucleotide degradation can also enter salvage pathways and then be reverted to complete nucleotides. We will now look at all these pathways in turn.
There are two major synthetic pathways, for purine and pyrimidine bases, respectively. Each pathway diverges towards its end to produce the two respective different bases. We will begin with the pathway for purine synthesis.
| 16.3 |
Purine synthesis |
| 16.3.1 |
Overview of purine synthesis |
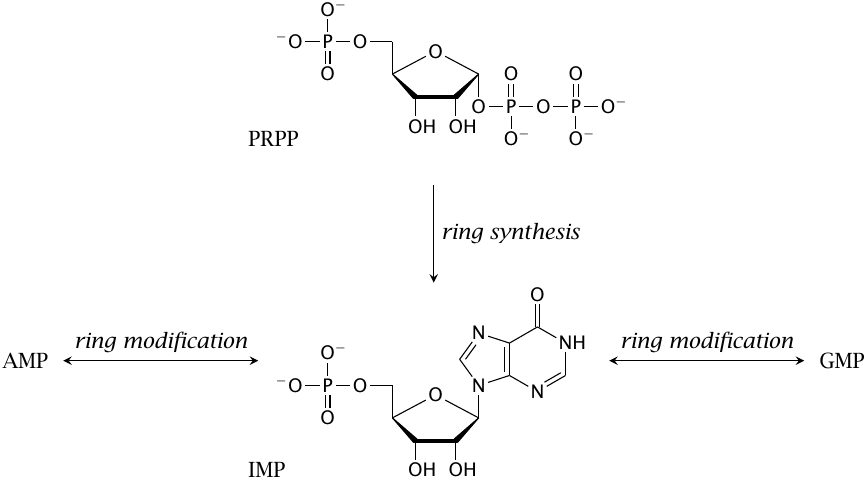
Adenine and guanine nucleotides are derived from a common precursor, inosine monophosphate (IMP), which consists of ribose phosphate and a purine derivative known as hypoxanthine. IMP is synthesized from 5′-phosphoribosyl-1′-pyrophosphate (PRPP) in a sequence of no less than 10 steps. In two of these steps, single carbon units are acquired from N10-formyltetrahydrofolate.
The conversions of IMP to AMP and to GMP, respectively, are reversible (but the reversal is mediated by different enzyme reactions). Therefore, AMP and GMP can be converted into one another via IMP as required.
| 16.3.2 |
IMP synthesis (1) |
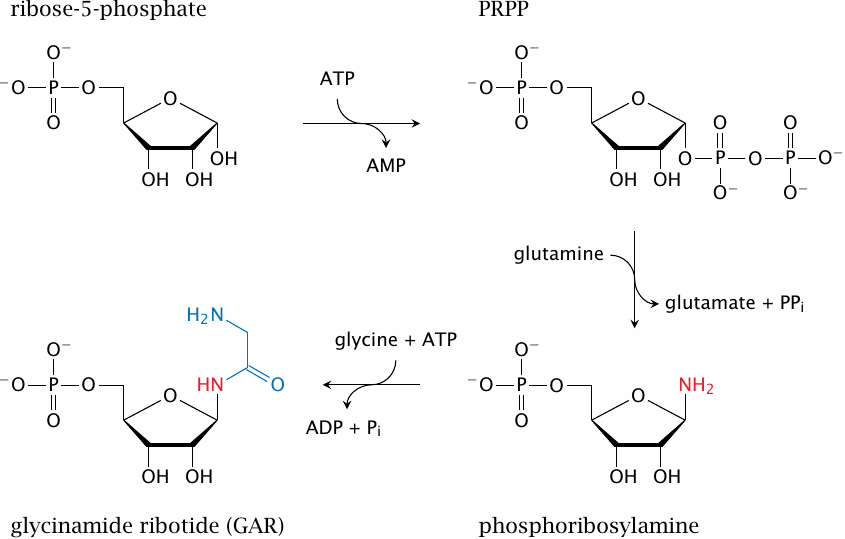
The substrate of the first reaction, ribose-5-phosphate, is supplied by the hexose monophosphate shunt. The enzymes for the reactions shown in this slide are, in order, ribose phosphate diphosphokinase, glutamine-PRPP-amidotransferase, and glycinamide ribotide synthetase.
| 16.3.3 |
IMP synthesis (2) |
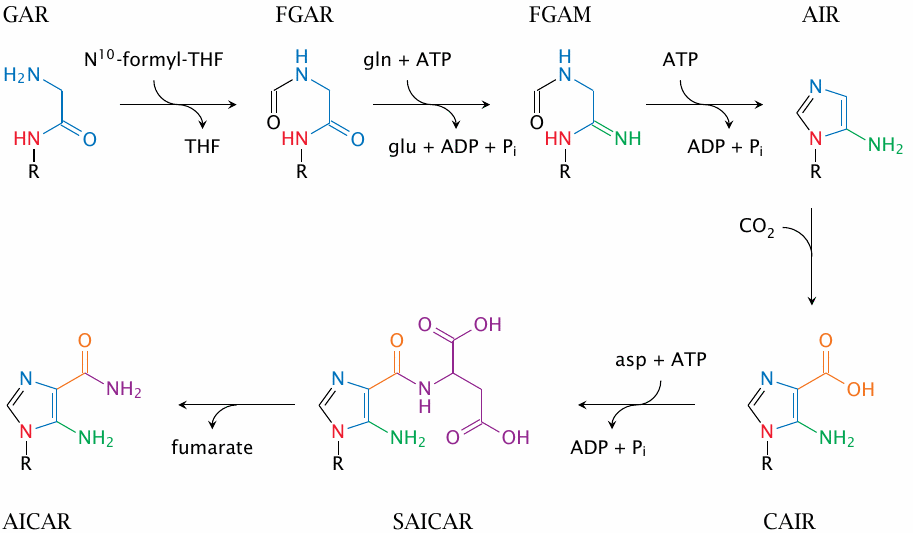
Construction of the hypoxanthine ring continues in a piecemeal fashion; atoms highlighted in different colors are derived from different precursors. Products (enzymes): FGAR, formylglycinamide ribotide (GAR transformylase); FGAM, formylglycinamidine ribotide (FGAM synthetase); AIR, 5′-aminoimidazole ribotide (AIR synthetase); CAIR, 4′-carboxy-5′-aminoimidazole ribotide (AIR carboxylase); SAICAR, succinyl-aminoimidazole-carboxamido-ribotide (SAICAR synthetase); AICAR, aminoimidazole-carboxamido-ribotide (adenylosuccinate lyase).
| 16.3.4 |
IMP synthesis (3) |
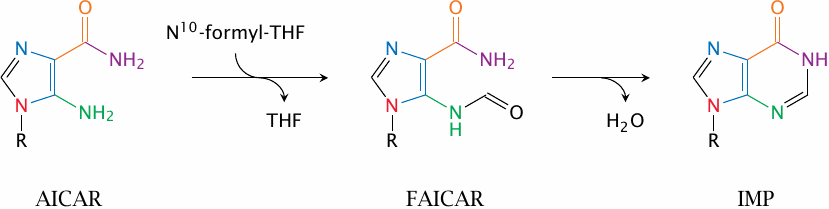
AICAR transformylase and IMP cyclohydrolase conclude the synthesis of the ribose-attached hypoxanthine ring.
Most of the chemistry in this pathway seems quite straightforward. Where ATP or GTP come into play, they are used to phosphorylate hydroxyl groups, which are thus activated for substitution by ammonia (released from glutamine) or by the amino group of aspartate.
The most remarkable reaction appears to be the direct carboxylation of 5-aminoimidazole ribotide (AIR) by AIR carboxylase, which unlike e.g. pyruvate carboxylase requires neither ATP nor biotin. Indeed, AIR carboxylase uses CO2 directly instead of ATP-activated bicarbonate, which is used by the biotin-dependent enzymes. This is made possible by its energetic coupling with the subsequent reaction (see next slide).
| 16.3.5 |
A bifunctional enzyme combines AIR carboxylase and SAICAR synthetase activities |
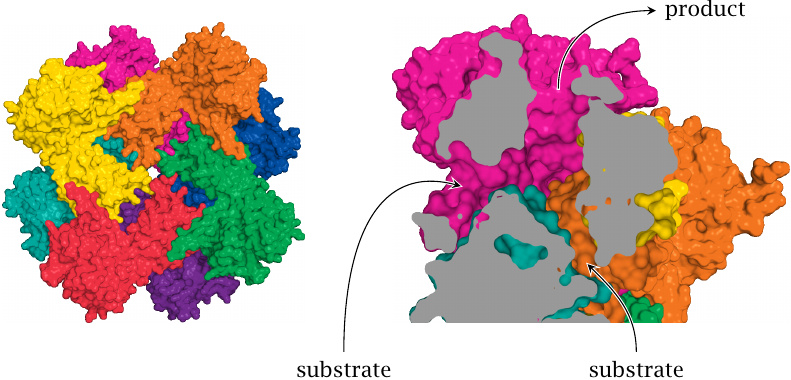
AIR carboxylase is actually part of a bifunctional enzyme that also contains the next enzyme activity in the pathway (SAICAR synthetase), and the carboxylated product (CAIR) is directly channeled to the SAICAR synthetase active site. The two active sites are located close to the entrances and the exit, respectively, of a connecting tunnel [110].
The equilibrium of the AIR carboxylase reaction probably does not favor the product (CAIR). In contrast, the SAICAR synthetase is driven forward by ATP hydrolysis. Thus, the tunnel that connects the two active sites would seem to function like a “vacuum cleaner” that removes all the product from the AIR carboxylate site and thereby pulls that reaction forward also. Structure rendered from 2h31.pdb.
| 16.3.6 |
Synthesis of AMP from IMP |
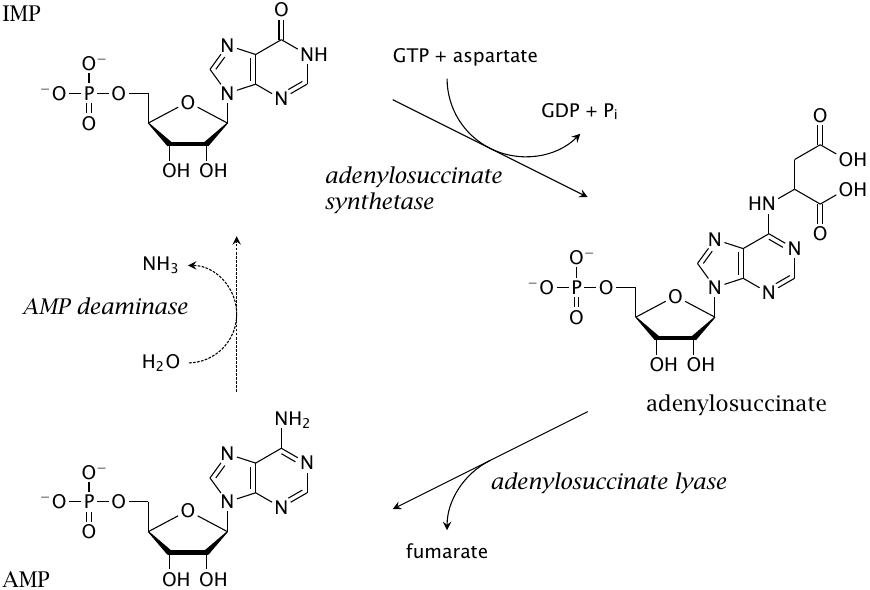
The synthesis of AMP from IMP occurs in two steps. In the first step, an aspartate is acquired to form adenylosuccinate. The subsequent elimination of fumarate—or, strictly speaking, the elimination of AMP from fumarate—is similar to the conversion of SAICAR to AICAR (slide 16.3.3). Indeed, both reactions are carried out by the same enzyme, which is named ‘adenylosuccinate lyase’ after the reaction depicted here.
If the supply of AMP exceeds demand, it can be converted back to IMP by AMP deaminase, either for degradation or for conversion to GMP (see slide 16.3.7). Taken together, the three reactions in this slide create a cycle that does not accomplish any net turnover of AMP but simply splits aspartate to fumarate, with the hydrolysis of GTP. This so-called purine nucleotide cycle may indeed be activated in muscle tissue, where it could serve to swiftly increase the pool of TCA cycle intermediates in times of increased energy demand. However, it seems that a genetic deficiency for AMP deaminase is quite common and usually asymptomatic [111], suggesting that the function of this cycle is not crucial for muscle function, although some divergent findings have been reported [112].
| 16.3.7 |
Synthesis of GMP from IMP |
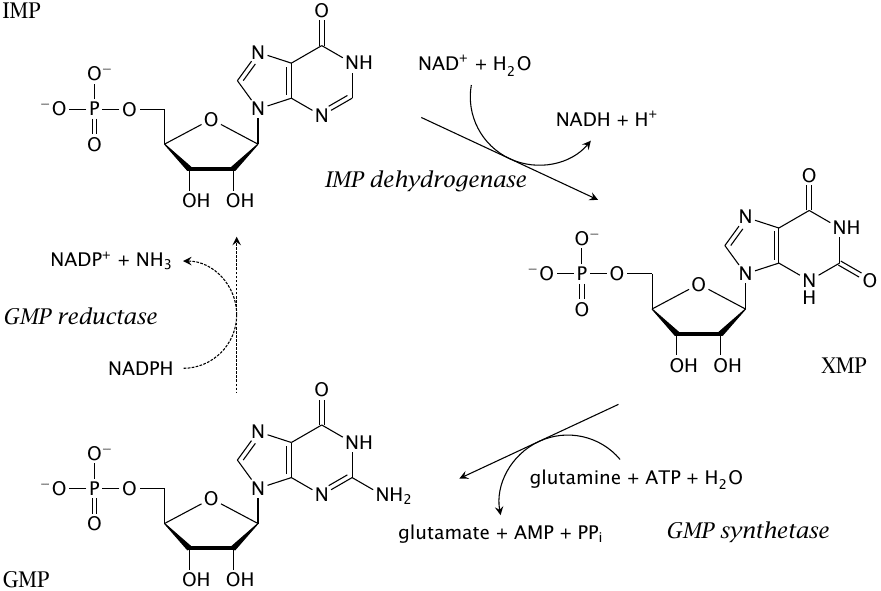
After the initial dehydrogenation of IMP to xanthosine monophosphate (XMP), glutamine serves once more as the nitrogen donor in the conversion of this intermediate to GMP. Excess GMP can be turned back into IMP for degradation or conversion to AMP.
Note the crosswise utilization of cosubstrates in the syntheses of AMP and GMP: AMP synthesis requires GTP, whereas GMP synthesis is driven by ATP. Therefore, an oversupply of either purine nucleotide will accelerate the synthesis of the other one. This arrangement helps to adjust the abundance of the two purine bases according to metabolic demands. Additional mechanisms that help to ensure this balance are discussed in the next slide.
| 16.3.8 |
Feedback regulation in purine synthesis |
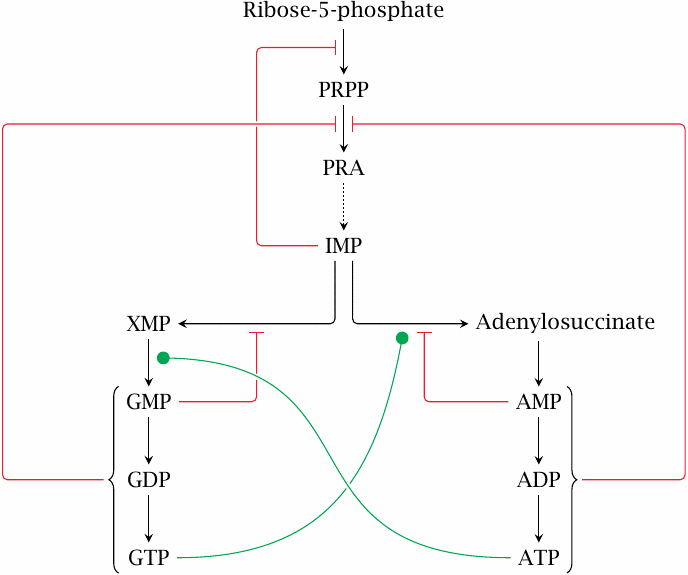
Purine synthesis is subject to feedback inhibition at several levels. All guanine and adenine nucleotides allosterically inhibit the synthesis of phosphoribosylamine (PRA) from PRPP. The common precursor IMP inhibits the formation of PRPP itself, as do ADP and GDP (not shown). These effects adjust the flow through the common pathway up to IMP in keeping with the total need for purine nucleotide synthesis.
The balance between AMP and GMP synthesis is maintained by the crosswise cosubstrate requirement that was discussed above (green lines). In addition, GMP and AMP both inhibit the formation of their immediate precursors from IMP; this inhibition is competitive.
| 16.4 |
Utilization of dietary nucleic acids |
| 16.4.1 |
Overview of digestion and utilization of nucleic acids |
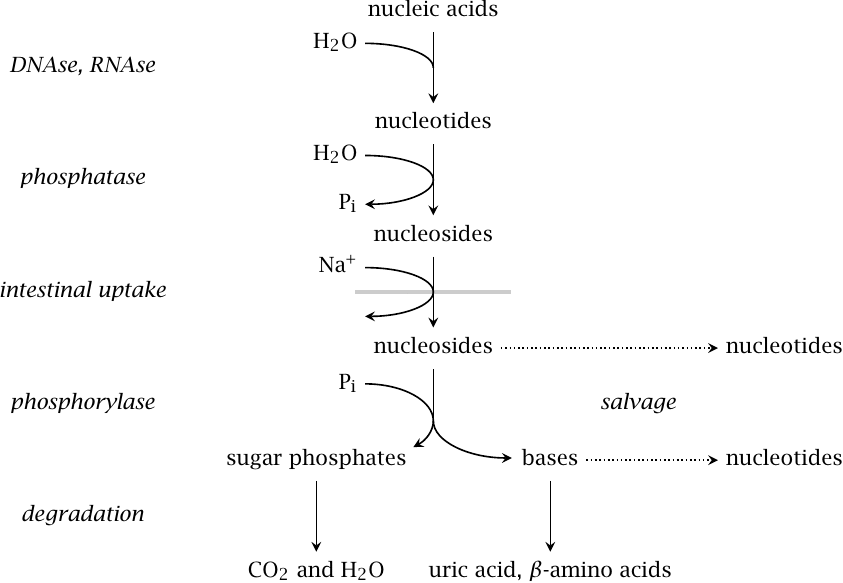
Nucleic acids are of nutritive value mainly because of the sugars they contain, namely, ribose and deoxyribose. In the small intestine, pancreatic nucleases (DNAse and RNAse) break down nucleic acids to nucleotides, which are dephosphorylated by alkaline phosphatase to nucleosides; the latter are taken up by sodium-coupled active transport. There are two intestinal transporters that prefer purine and pyrimidine nucleosides, respectively, but have fairly broad and overlapping specificity. This loose specificity permits piggyback uptake of nucleoside analogue drugs such as idoxuridine and 6-mercaptopurine riboside (see section 16.9).
After uptake, nucleosides mostly undergo degradation in the liver. Cleavage by purine and pyrimidine nucleoside phosphorylases gives free bases and ribose- or deoxyribose-1-phosphate. The sugar phosphates are converted to mainstream degradative intermediates via short adapter pathways (see next slide).
Both free bases and uncleaved nucleosides can in principle be reused toward the synthesis of nucleotides. However, the salvage pathways that accomplish this appear to mostly process endogenously synthesized bases, whereas those obtained from the diet mostly undergo degradation and excretion [113,114].
| 16.4.2 |
Utilization of ribose and deoxyribose |
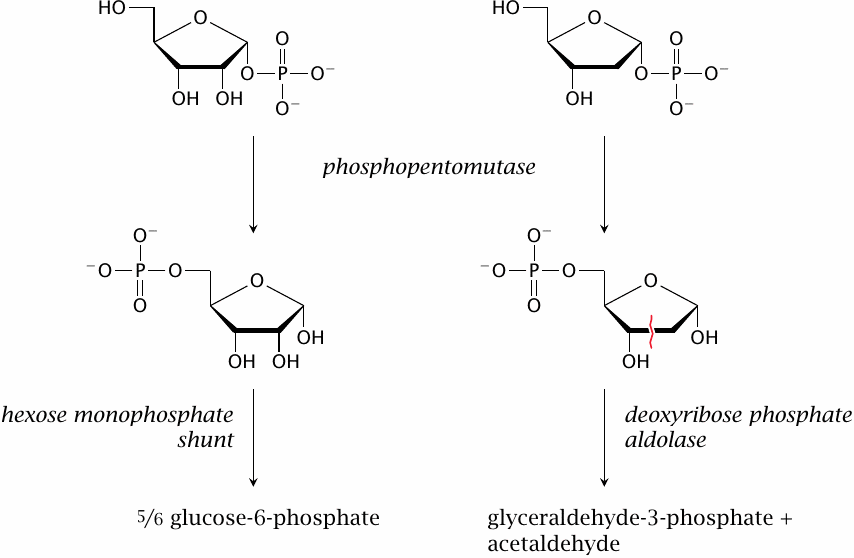
The ribose-1-phosphate and deoxyribose-1-phosphate that are released from dietary nucleosides are first converted to the respective 5-phosphates by phosphopentomutase. This reaction is analogous to the interconversion of glucose-6-phosphate and glucose-1-phosphate by phosphoglucomutase (see slide 8.3.1).
The subsequent utilization pathways diverge. Ribose-5-phosphate is an intermediate of the hexose monophosphate shunt and can be converted to glucose-6-phosphate in this pathway (see slide 9.2.2). Deoxyribose-5-phosphate is cleaved—across the red squiggly line in the figure—by a specific aldolase, which produces glyceraldehyde-3-phosphate, an intermediate of glycolysis, and acetaldehyde. The latter is also an intermediate in ethanol degradation (see slide 7.4.2) and is utilized along this pathway.
| 16.5 |
Purine degradation and salvage pathways |
The following pathways account for the degradation of endogenous purine nucleotides, as well as of the free bases that result from the phosphorolysis of dietary ones.
| 16.5.1 |
Degradation of endogenous purine nucleotides (overview) |
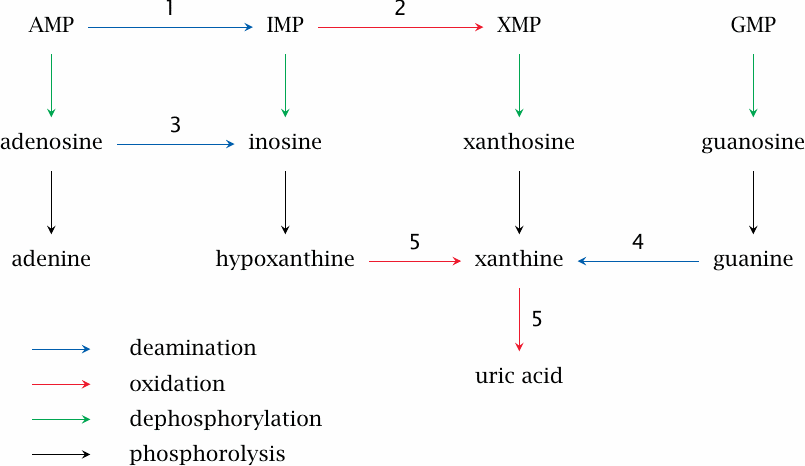
Enzymes: 1, AMP deaminase; 2, IMP dehydrogenase; 3, adenosine deaminase; 4, guanase; 5, xanthine dehydrogenase or oxidase.
The AMP deaminase reaction is shown in the next slide; the adenosine deaminase reaction is analogous. The guanase and xanthine dehydrogenase/oxidase reactions are depicted in slide 16.5.3. The dephosphorylation of the monophosphate nucleotides to the nucleosides and the subsequent phosphorolysis to free bases occurs in the same way as outlined above for dietary nucleotides and nucleosides (slide 16.4.1).
| 16.5.2 |
Adenine nucleotide degradation |
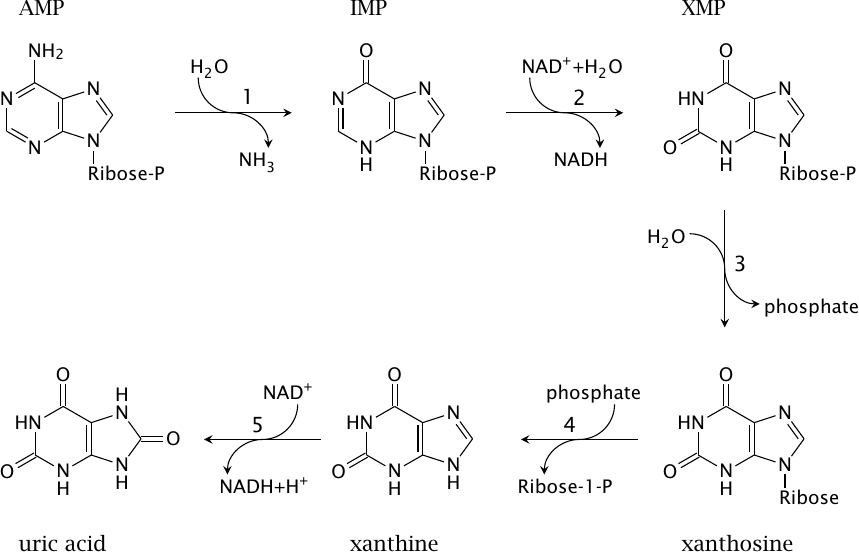
As shown in slide 16.5.1, the degradation of AMP can follow several alternative routes that perform dephosphorylation, phosphorolysis, deamination, and oxidation reactions in different sequences. This figure depicts one of these alternative routes in detail. The depicted reactions are catalyzed by AMP deaminase (1), IMP dehydrogenase (2), 5′-nucleotidase (3), purine nucleoside phosphorylase (4), and xanthine dehydrogenase/oxidase (5).
| 16.5.3 |
The guanase and xanthine dehydrogenase/oxidase reactions |
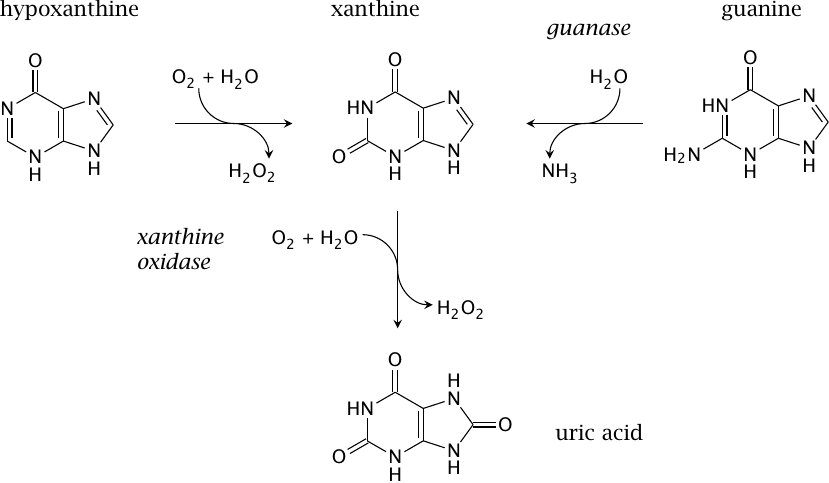
Guanase deaminates guanine to xanthine, which is again converted to uric acid by xanthine dehydrogenase. The latter enzyme also occurs in a second variation, which arises through protein modification. Intracellularly, most enzyme molecules function as a dehydrogenase and reduce NAD+, as shown in the previous slide. Partial proteolysis, or the introduction of a strategic disulfide bond, prevent the interaction of the enzyme with NAD+, and the molecule then switches to O2 reduction, as shown in this slide. The oxidase activity is found in extracellular enzyme molecules, which lack a supply of NAD+ anyway.
As we had seen before, adenosine degradation may produce free hypoxanthine. Xanthine dehydrogenase or oxidase both can convert hypoxanthine to xanthine and the latter to uric acid.
| 16.5.4 |
Renal urate elimination: tubular reuptake and secretion |
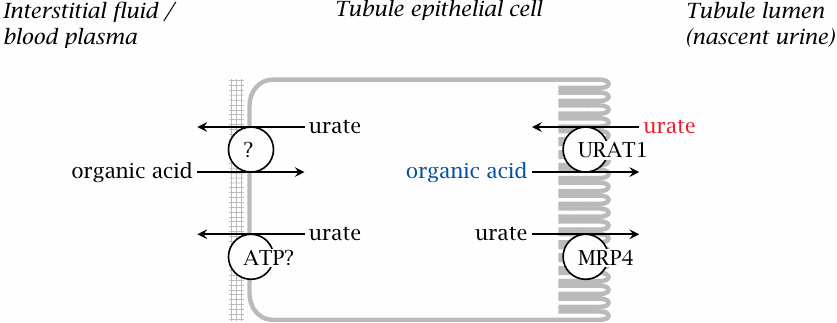
Most of the uric acid formed by purine degradation is eliminated via the kidneys. Urate is subject primarily to glomerular filtration and tubular reuptake (see slide 14.2.5), while tubular secretion (by an ABC transporter named MRP4) is less important.
Reuptake of urate from the primary filtrate is mediated by the URAT1 exchange transporter. Several drugs and metabolites that affect renal urate elimination interact with this transporter [115]. Typically, when present in the lumen of the tubule, such compounds will compete with urate for reuptake; this is the mode of action of uricosuric drugs like benzbromarone or probenecid. On the other hand, when present inside the epithelial cells, they may act as substrates for exchange and therefore increase the rate of reuptake, as is the case with pyrazinoic acid (see slide 16.6.5).
| 16.5.5 |
Non-primates break down uric acid to allantoin |
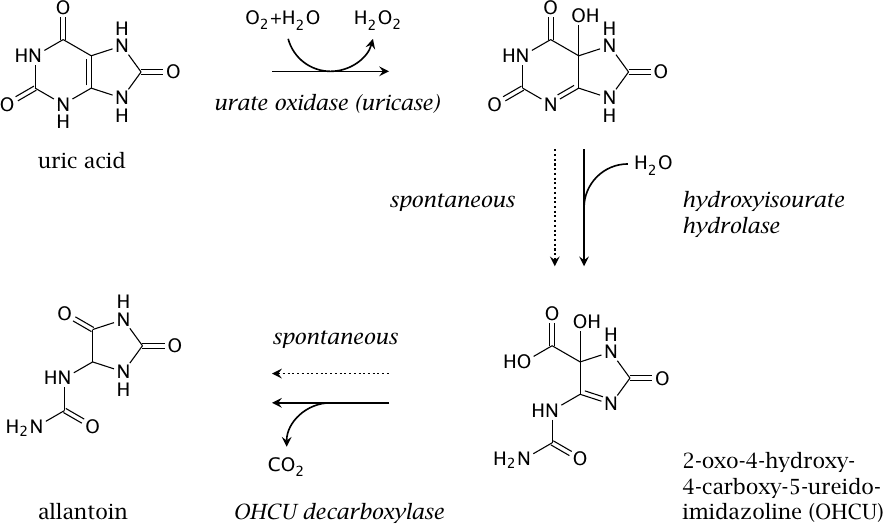
While uric acid is the terminal product of purine degradation in humans and other apes, many other organisms, ranging from fungi to mammals, perform several subsequent reactions that degrade uric acid further to allantoin, which is then excreted. Allantoin is more water-soluble than uric acid and therefore avoids the problems that can arise from the low solubility of uric acid in humans (see slide 16.6.2).
The first step in urate degradation is carried out by urate oxidase (uricase). Interestingly, humans actually possess an inactive copy of the urate oxidase gene, which indicates that at some point during evolution this enzyme activity was lost. Since the genetic enzyme defect spread throughout the entire population, there likely was some selective advantage to it. Several hypotheses have been proposed as to the nature of this advantage, ranging from antioxidant effects of uric acid over protection from UV irradiation to a rise in blood pressure. While the correlation between uric acid blood levels and hypertension is worth noting from a clinical point of view [116], the biological advantage of an increase in blood pressure is not obvious to me; and even if it existed, it would seem implausible for evolution to cripple a perfectly fine pathway in order to adjust the blood pressure when a plethora of other physiological regulation mechanisms for the same purpose was already available. Among the alternatives listed, the antioxidant role of uric acid is the best supported by the evidence (see Chapter 18).
While the role of urate oxidase has been known for a long time, the subsequent two enzyme reactions shown in this slide have only recently been discovered [117]. The reason for their delayed discovery is that urate oxidase alone is sufficient for the conversion of urate to allantoin, since the subsequent reactions also occur spontaneously. The major difference is that the spontaneous decay produces allantoin as a racemic mixture, whereas enzymatic formation gives rise to one enantiomer only.
| 16.5.6 |
Overview of purine salvage reactions |
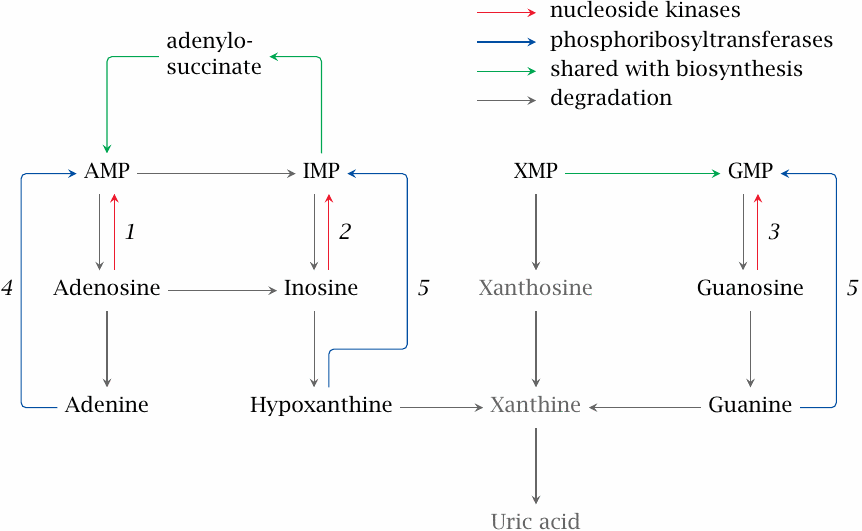
Nucleosides and free bases that arise during the degradation of nucleic acids and nucleotides can be salvaged and reused. This scheme shows the reactions involved in purine salvage, which produce AMP, GMP and IMP. The conversion of the latter to AMP or GMP (blue arrows) involves the same reactions as in de novo synthesis (see slides 16.3.6 and 16.3.7).
Enzymes: 1, adenosine kinase; 2, inosine kinase; 3, guanosine kinase; 4, adenine phosphoribosyltransferase (APRT); 5, hypoxanthine-guanine phosphoribosyltransferase (HGPRT). The APRT and HGPRT reactions are analogous to the orotate phosphoribosyltransferase reaction (see slide 16.7.1).
| 16.6 |
Diseases related to purine degradation |
| 16.6.1 |
Enzyme defects in purine degradation and salvage |
| Enzyme | Biochemical effects | Clinical symptoms |
| adenosine deaminase | accumulation of dA and dATP | severe combined immunodeficiency (SCID) |
| HGPRT | defective purine salvage, increased de novo synthesis and degradation | gout; impeded cerebral development and self-mutilation (Lesch-Nyhan syndrome) |
Adenosine deaminase deficiency induces apoptosis in T cells, which results in severe combined immunodeficiency (SCID; see section 20.2).
HGPRT is encoded on the X chromosome; therefore, the enzyme defect, which is known as Lesch-Nyhan syndrome, is observed mostly in boys. Excessive urate production and gout are expected, but the causation of the neurological symptoms is not well understood. Excessive activation of adenosine receptors in the brain during fetal development has been proposed. Disruption of dopamine activity in the brain has been repeatedly observed [118]; an interesting animal model of this effect has been described [119]. However, a clear picture of how HGPRT deficiency causes cerebral dopamine deficiency during development has not yet emerged.
| 16.6.2 |
Gout |
- Genetic or dietary factors cause chronically increased urate production or retention
- Urate has limited solubility and may form crystalline deposits, preferentially in joints and soft tissue
- Urate crystals activate inflammation and lead to arthritis that is painful and, in the long run, destructive
The mechanism that links urate crystals to inflammation has been elucidated [72] and is similar to the one that applies to cholesterol crystals [71].
| 16.6.3 |
Diets and drugs that may promote gout |
- too much food, too rich in purines
- excessive fructose or sucrose
- alcoholic beverages—but not all kinds: beer yes, wine no
- anorexia nervosa (!)
- drugs that interfere with uric acid secretion: pyrazinamide, salicylic acid
- drugs that contain purines: dideoxyadenosine
It seems that the uptake of purines and the subsequent production of uric acid are linearly related to purine ingestion [114]. Therefore, a purine-rich diet is a straightforward way to contract gout.
There are several plausible connections between alcohol and gout, of which no single one has unequivocally been shown to be the dominant one. Degradation of alcohol via alcohol dehydrogenase and then aldehyde dehydrogenase produces NADH, which shifts the equilibrium of the lactate dehydrogenase reaction from pyruvate to lactate (see slide 7.4.2). Lactate may increase the retention of uric acid in the kidneys by serving as an exchange substrate in tubular transport (see slide 16.5.4).
A statistical study found an association between gout and the consumption of beer, but not of wine [120]. The amounts of alcohol ingested by both groups were similar, suggesting that moderate consumption of alcohol as such does not promote gout. Beer is higher in calories and in purines than wine, which may account for the difference observed in this study.
Considering the association of a rich diet with gout, it seems surprising that anorexia nervosa, an eating disorder in which patients eat only the bare minimum required to ward off death, and sometimes less, may also lead to gout [121]. This has been ascribed to the formation of ketone bodies, which as organic acids may also serve as exchange substrates in the tubular reuptake of uric acid and thereby increase urate retention.
The drug 2,3-dideoxyadenosine inhibits retroviral reverse transcriptase and is used in the treatment of HIV infections (see slide 16.9.11). It undergoes degradation like other purine nucleotides and as such will contribute to uric acid production, which may occasionally trigger gout [122].
| 16.6.4 |
Gout: the fructose connection |
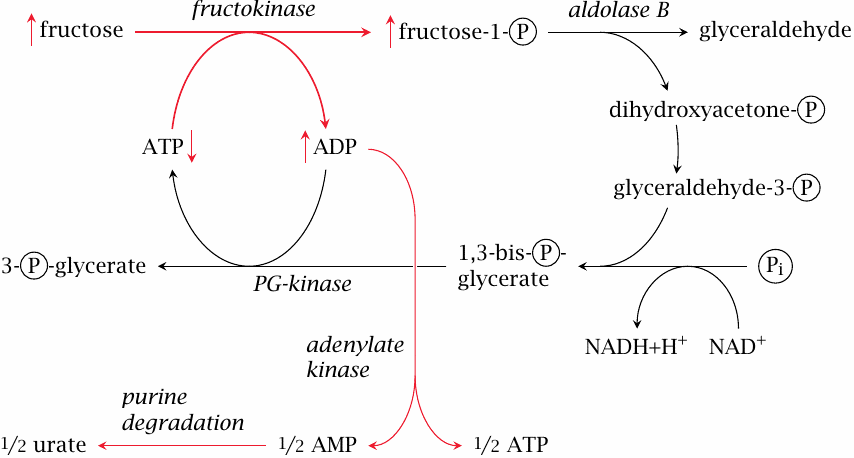
Fructose has been linked to increased uric acid production both experimentally [123] and statistically [124]. The mechanism is as follows: Fructokinase produces fructose-1-phosphate more rapidly than it can be turned over by aldolase B. As a result, phosphate is tied up and no longer available for the regeneration of ATP. This raises the level of ADP, which is disproportionated by adenylate kinase to ATP and AMP, and the latter enters degradation via AMP deaminase (to IMP) or 5′-nucleotidase (to adenosine; see slide 16.5.1).
In keeping with the assumption that uric acid formation is promoted by limiting activity of aldolase B, heterozygous carriers of a deficient gene for this enzyme experience a greater increase of urate synthesis in response to a fructose challenge than individuals with two intact gene copies [125]. As noted before, a homozygous defect of aldolase B causes hereditary fructose intolerance. In this condition, phosphate sequestration and ATP depletion reach a much greater extent, with much more immediate and disastrous consequences for the liver cells (see slide 4.2.2).
| 16.6.5 |
Drugs that affect purine degradation and elimination |
Allopurinol is an inhibitor of xanthine dehydrogenase/oxidase. When it is used, hypoxanthine and xanthine will accumulate instead of uric acid. These intermediates are more soluble than uric acid and are readily excreted. Allopurinol is very effective and a mainstay in the treatment of gout.
Probenecid and benzbromarone are uricosuric drugs, that is, they increase the excretion of uric acid with the urine through inhibition of its tubular reuptake by the URAT1 transporter (see slide 16.5.4). The increased efficiency of urate excretion lowers the urate levels in blood and tissues, which in turn reduces the risk of precipitation in the joints. Uricosuric drugs also are widely used in gout therapy.
Some drugs or drug metabolites promote the tubular reuptake of uric acid, presumably by functioning as exchange substrates at the URAT1 transporter. This effect can occur with salicylic acid, and it is very pronounced with pyrazinoic acid and its 5-hydroxy derivative, the metabolites of the tuberculostatic drug pyrazinamide. Treatment with pyrazinamide, which is carried out over several months and at dosages of gram amounts per day, may therefore trigger gout.
| 16.6.6 |
Acute urate nephropathy in tumor lysis syndrome |
- Occurs during chemotherapy of malignancies, particularly with lymphomas and leukemias
- Chemotherapy causes acute decay of large numbers of tumor cells
- Degradation of nucleic acids from decaying cells produces large amounts of uric acid
- Uric acid in nascent urine exceeds solubility and precipitates, clogging up and damaging the kidney tubules
- Clinically manifest as acute kidney failure with high fatality rate
The underlying problem in acute urate nephropathy is again an excess of uric acid, but in this case it occurs very suddenly and on an altogether different scale than in simple gout. As a consequence, the capacity for reuptake in the proximal tubules is overwhelmed, and the excess uric acid appears in large quantities in the distal tubules. As water is reclaimed from the nascent urine, the uric acid becomes more concentrated. Moreover, the pH value in the distal tubule is often acidic, which will cause uric acid (pKa =5.75) to become protonated and precipitate.
The condition is particularly common with lymphomas and leukemias. Unlike most other cancers, in which most tumor cells are contained in a single solid mass that is surgically removed prior to chemotherapy, leukemias and most lymphomas grow diffusely and are not amenable to surgical removal; therefore, chemotherapy is used right from the start and impinges on a far greater number of malignant cells in the body. As a preventive measure, leukemia or lymphoma patients that undergo chemotherapy receive allopurinol concomitantly. This treatment has greatly reduced the incidence of acute urate nephropathy.
| 16.6.7 |
Rasburicase, a better preventive treatment for urate nephropathy |
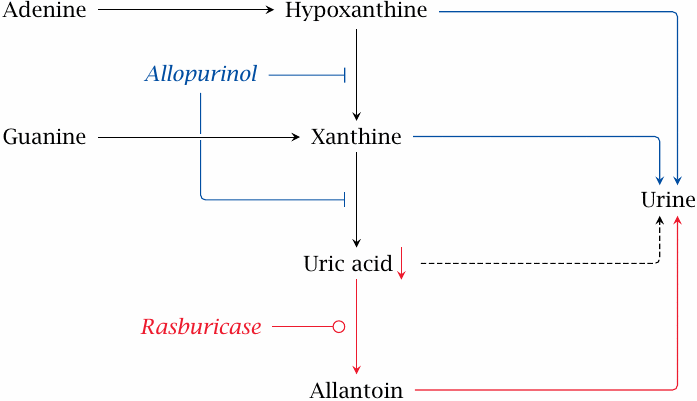
This slide shows the action modes of allopurinol and of rasburicase, which is an uricase enzyme from an Aspergillus mold that is recombinantly produced in baker’s yeast (Saccharomyces cerevisiae). The enzyme will convert uric acid to allantoin (see slide 16.5.5). This mode of action complements the decreased formation of uric acid attained with allopurinol. Like the latter drug, rasburicase is used—by way of intravenous infusion—to prevent acute urate nephropathy in leukemia patients undergoing chemotherapy. Individually, rasburicase is superior to allopurinol, but it should also be possible to use both drugs in combination.
Since rasburicase is a non-self protein, it is immunogenic, and it thus may induce the formation of antibodies that inactivate it and additionally may cause allergic complications. This problem is mitigated by the immunosuppressive effect of chemotherapy and, in leukemia, of the disease itself. However, it will likely make rasburicase unsuitable for long-term use in gout patients [126].
| 16.7 |
Synthesis and degradation of pyrimidines |
| 16.7.1 |
Synthesis of pyrimidines (1) |
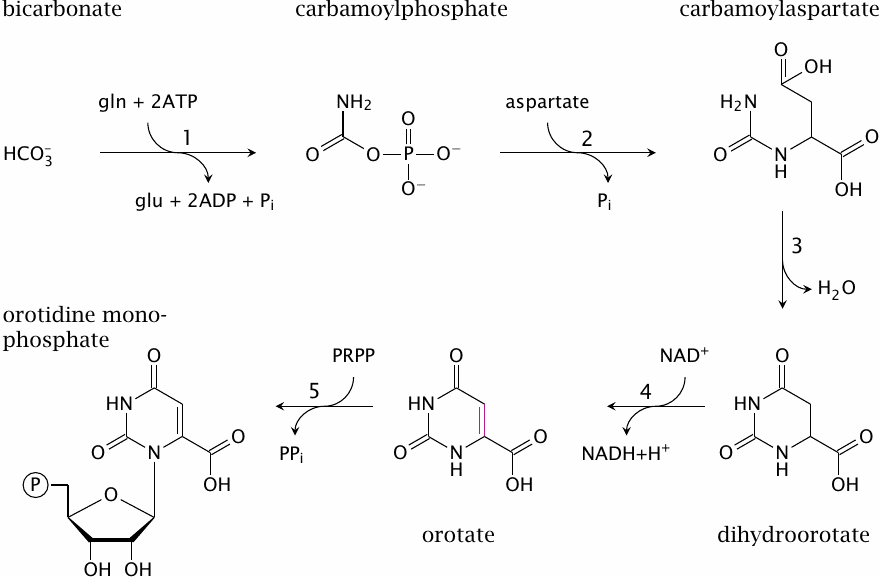
In contrast to the purine rings of AMP and GMP, which are assembled atop the phosphoribose moiety, the skeleton of the pyrimidine bases is synthesized before attachment to ribose phosphate.
The first intermediate is carbamoylphosphate. The corresponding enzyme, carbamoylphosphate synthetase 2, uses glutamine as the nitrogen donor; in contrast, carbamoylphosphate synthetase 1, which occurs in the urea cycle, uses free ammonia directly (see slide 12.3.1).
In the second reaction, aspartate transcarbamylase transfers the carbamoyl group of carbamoylphosphate to the α-amino group of aspartate, which yields carbamoylaspartate. Ring closure through intramolecular Schiff base formation (3) yields dihydroorotate, and subsequent dehydrogenation (4) produces orotate. Orotate phosphoribosyltransferase (5) forms orotidine-5′-monophosphate (OMP), in a reaction that resembles purine salvage (compare slide 16.5.6). Indeed, this enzyme has relatively broad specificity and also functions in the salvage of uracil and in the activation of its analogue 5-fluorouracil (see slide 16.9.1).
| 16.7.2 |
Synthesis of pyrimidines (2) |
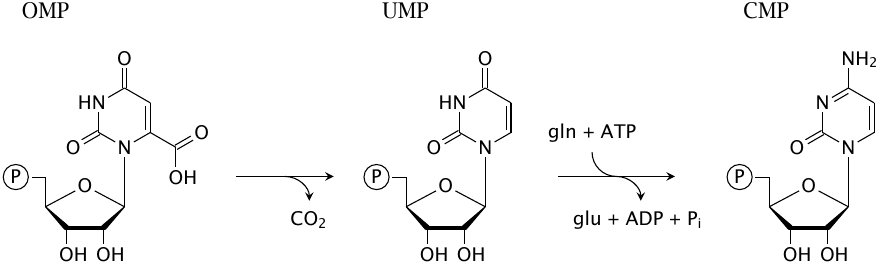
From OMP, it is only a short way to the final pyrimidine nucleotides. Decarboxylation of OMP yields UMP directly; glutamine donates another nitrogen in the synthesis of CMP from UMP.
| 16.7.3 |
Degradation of pyrimidines |
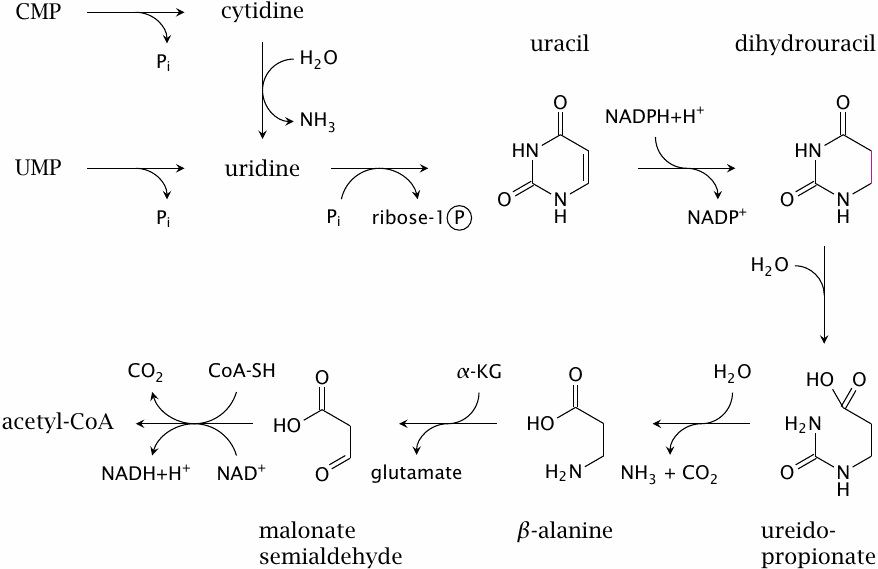
Like their synthesis, degradation of pyrimidine bases is also fairly straightforward. Cytidine is deaminated to uridine. Pyrimidine nucleoside phosphorylase cleaves uridine to free uracil and ribose-1-phosphate. One of the amide bonds in the uracil ring is opened through hydrolysis, and ammonia and CO2 are cleaved off to produce β-alanine. This compound may be excreted with the urine, or it may be transaminated to malonyl semialdehyde and converted to acetyl-CoA.100
Thymine is degraded in the same manner as uracil, producing methylated analogues at each step along the way. In the final step, methylmalonyl-semialdehyde gives rise to propionyl-CoA, which is utilized as outlined in slide 10.3.6.
| 16.7.4 |
β-Alanine may be used to synthesize carnosine |
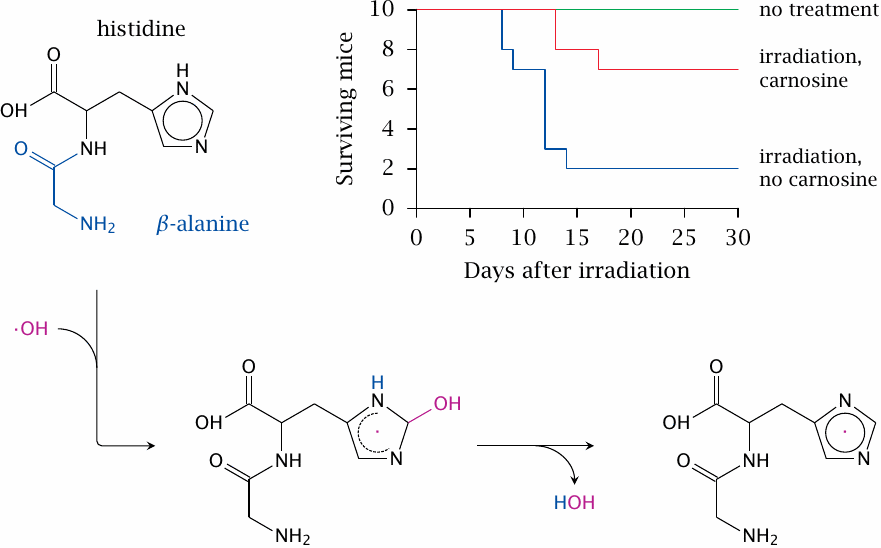
Carnosine is a dipeptide of β-alanine and histidine. It is found in high concentrations in muscle (~2 mM in humans, but higher in many other mammals [127]); lower concentrations are found in nerve cells. One intriguing property is illustrated here, namely, its reaction with hydroxyl radicals, which results in the formation of a more stable carnosine radical [128]. Hydroxyl radicals are produced by ionizing radiation and mediate most of the cellular damage caused by radiation. The animal experiment depicted in the plot [129] suggests that carnosine may scavenge those radicals and reduce the inflicted damage. Note though that the number of mice used in this experiment was rather small.
While numerous biological activities have been proposed for carnosine, the most important one is likely to buffer the lactic acid that accumulates in skeletal muscle during anaerobic exercise. The pKa value of carnosine is very close to the physiological intracellular pH, which maximizes buffer capacity. Carnosine and its methylated analogue ophidine are highly abundant (40–50 mM) in the muscle tissue of whales. This large buffer capacity likely helps the whales to sustain extended periods of anaerobic muscle metabolism during their dives to the deep [127].
| 16.8 |
Synthesis of deoxyribonucleotides |
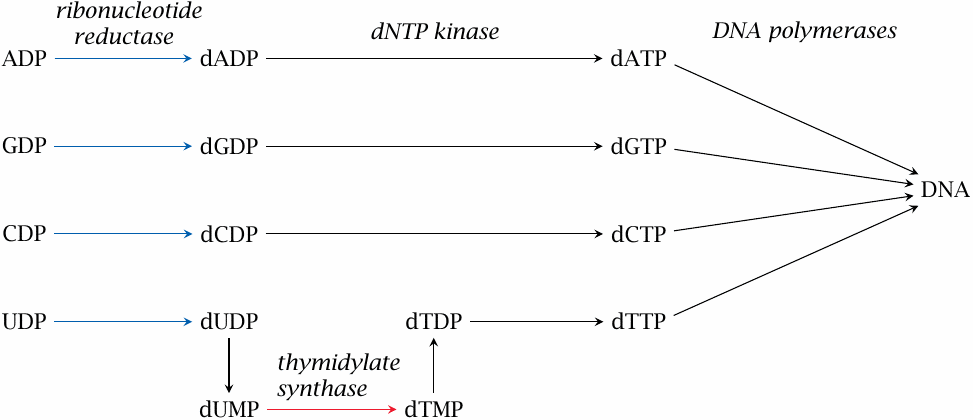
The synthetic pathways covered so far all produce ribonucleotides. In addition, we also need deoxyribonucleotides as precursors for DNA synthesis. These are made from ribonucleotides.
The conversion of ribonucleotides to deoxyribonucleotides occurs at the level of the nucleoside diphosphates. All four ribonucleotides—ADP, CDP, GDP and UDP—are reduced by the same enzyme, ribonucleotide reductase (RR). This reaction requires NADPH.
With dA, dC and dG, the deoxytriphosphates are then obtained simply through ATP-dependent phosphorylation by nucleoside diphosphate kinase. In contrast, uracil still has to be converted to thymine for the purpose of DNA synthesis. In order to prevent incorporation of uracil into DNA, dUDP is first dephosphorylated to dUMP by a cognate phosphatase. dUMP then enters the thymidylate synthase reaction (TS); the product, dTMP, is phosphorylated twice to produce dTTP.
| 16.8.1 |
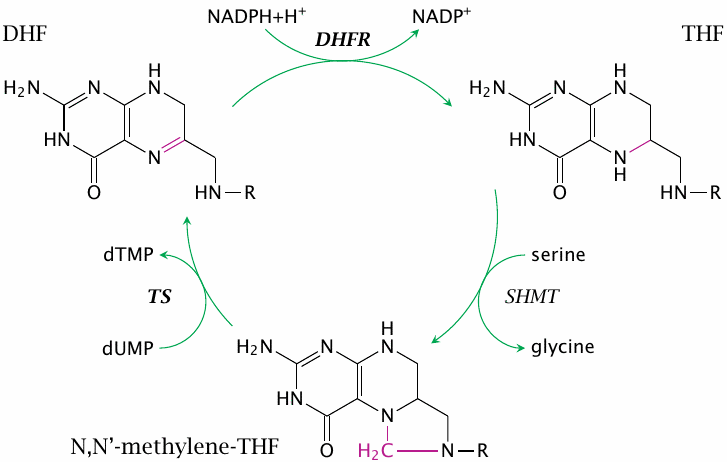
The thymidylate synthase reaction |
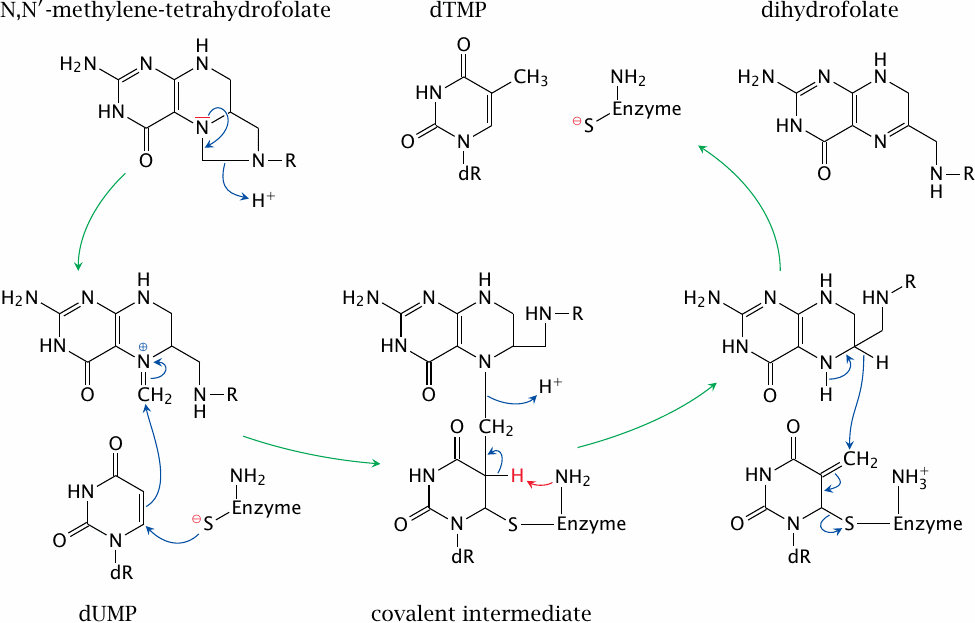
Thymidylate synthase acquires a single carbon from the cosubstrate N,N’-methylene-tetrahydrofolate (methylene-THF) and transfers it to dUMP. The reduction of the methylene carbon to the methyl group as found in the product (dTMP) occurs at the expense of THF, which is thereby converted to dihydrofolate and has to be reduced again to THF by dihydrofolate reductase (see slide 15.2.2).
Note that, halfway through the reaction, the enzyme, the substrate, and the cosubstrate are all joined together in a covalent intermediate, which is then resolved through the abstraction of a proton from position 5 of the uracil by a catalytic base in the enzyme’s active site (red arrow). This mechanism forms the basis of the inhibition of thymidylate synthase by the antitumor drug 5-fluorouracil (see slide 16.9.1).
| 16.9 |
Nucleotide antimetabolites as anticancer and antiviral drugs |
| Therapeutic principle | Examples |
| direct inhibition of DNA/RNA polymerization | dideoxyadenosine, cytosine arabinoside, acyclovir |
| inhibition of nucleotide synthesis | mercaptopurine, fluorouracil, methotrexate |
| Incorporation of mutagenic analogues into DNA | idoxuridine |
Nucleotide analogues are of major importance in the chemotherapy of both cancers and virus infections. With antiviral drugs, selective toxicity for virus-infected cells is possible, because viral enzymes often differ from the analogous human ones with respect to their susceptibility to specific inhibitors. In contrast, the same kind of selectivity is not possible with cancer cells, which have the same enzymes as noncancerous cells do; antimetabolites that kill cancer cells are invariably toxic to noncancerous cells also. However, cancer cells are often more susceptible to these cytotoxic drugs because they are more readily driven by them into apoptosis, that is, programmed cell death (see slide 19.5.1), which is triggered by various forms of DNA damage.
As will become clear from the discussion of individual examples below, some drugs can cause interference by more than one mechanism.
| 16.9.1 |
Dual action mode of 5-fluorouracil |
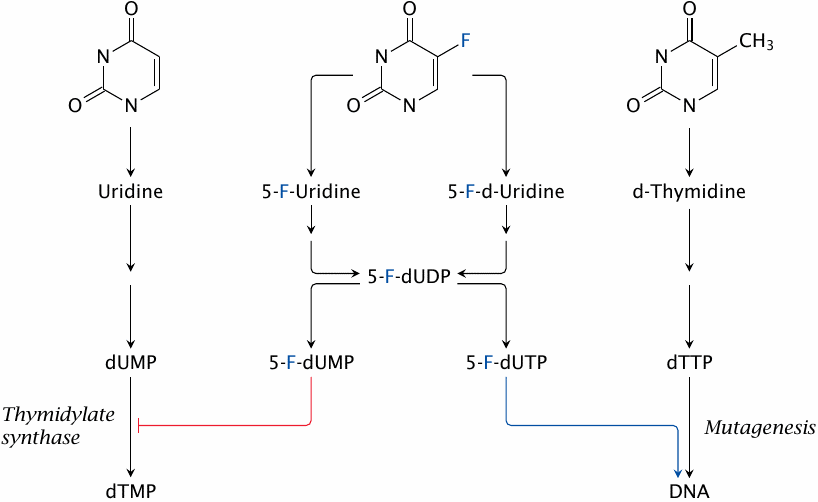
5-fluorouracil (5-FU) is a base analogue that mimics both uracil and thymine. The dual resemblance endows it with a twofold mode of metabolic activation and of action. One of several initial activation products, 5′-fluorodeoxyuridine, is also used as a drug itself.
The metabolic activation of 5-FU occurs along the so-called salvage pathways that recycle bases and nucleosides released by nucleic acid degradation. 5-FU is activated by the same enzymes that salvage uracil. A key activation product is 5-FdUMP, which is a suicide substrate for thymidylate synthase (see next slide). This is its major mode of action and the one that makes it an antimetabolite.
5-FU is also activated by enzymes that salvage thymine and thus may wind up as 5′-fluoro-dUTP, which is incorporated into DNA; this will cause point mutations (see slide 16.9.3).
| 16.9.2 |
Inhibition of thymidylate synthase by 5-fluorouracil |
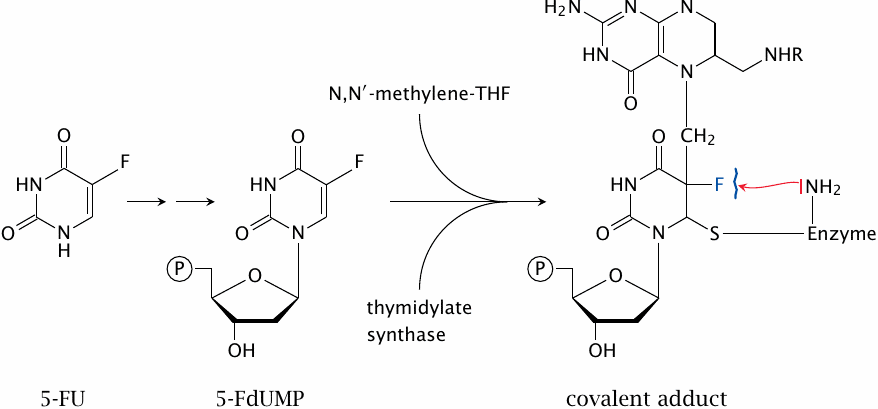
5′-Fluorouridine monophosphate (5-FdUMP) is accepted by thymidylate synthase as a substrate and reacts with methylene-THF and the enzyme right up to the point where all three are joined in a covalent intermediate (slide 16.8.1). At this stage, however, the fluorine in position 5 of the uracil resists abstraction by the catalytic base of the enzyme. The covalent intermediate therefore cannot be resolved, and the enzyme molecule remains irreversibly locked up.
Inhibition of thymidylate synthase will starve the cell of thymine required for DNA synthesis. Fluorouracil has shown particularly good activity in colon cancer, but there seems to be no known biochemical reason to account for this empirical rule.
| 16.9.3 |
Mutagenesis through mispairing of the 5-FU iminol tautomer |
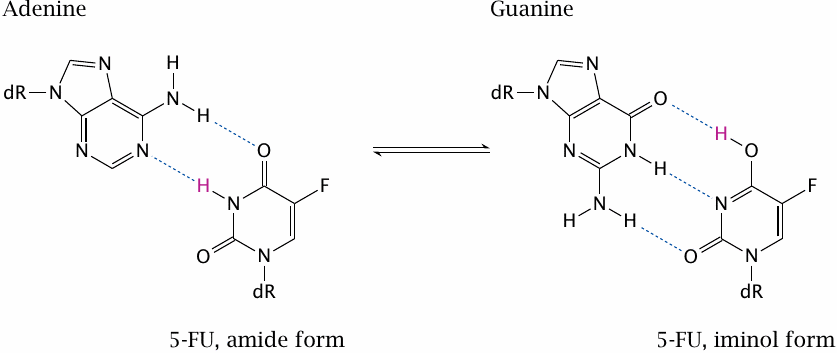
5-F-dUTP can be incorporated into DNA and promote point mutations. This is due to the fluorine, which withdraws electrons from the ring; this, in turn, pulls electrons into the ring from other substituents and encourages the molecule to assume the iminol configuration. In this configuration, the base no longer pairs with adenine but with guanine.
If the iminol configuration is present during DNA replication, guanine will be chosen for incorporation into the opposite strand. This mutagenic effect of 5-FU augments its anticancer effect.
| 16.9.4 |
Thymine and various halogen analogues |
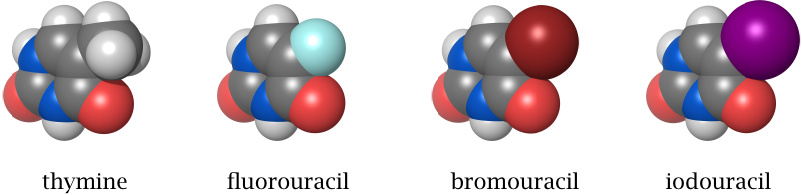
The bromo- and iodo-analogs of deoxythymidine, 5-bromouracil-deoxyriboside (5-BUdR) and 5-iodouracil-deoxyriboside (idoxuridine), are also incorporated into DNA. Bromine and iodine are larger than fluorine and similar to a methyl group in size. Therefore, the activated triphosphates more closely resemble dTTP and are more effectively incorporated than the fluoro-derivative.
Like fluorouracil, idoxuridine is used in tumor therapy. 5-BUdR is not used clinically but has in the past been widely used for shotgun mutagenesis experiments in genetic research.
| 16.9.5 |
Indirect inhibition of thymidine synthesis by methotrexate |
Methotrexate is an inhibitor of dihydrofolate reductase (see slide 15.2.2). This enzyme is required to reduce the dihydrofolate produced by the thymidylate synthase reaction to THF again, so that it can accept another carbon from serine hydroxymethyltransferase or another enzyme.
Methotrexate can be used in tumor therapy. Because it is not a nucleotide analogue and does not react with DNA, it is not mutagenic, which makes it more suitable than most base analogues in antiproliferative therapy not related to tumors. In particular, methotrexate is used to suppress lymphocyte proliferation in severe autoimmune diseases such as rheumatoid arthritis or Crohn’s disease.
| 16.9.6 |
Mercaptopurine inhibits purine synthesis |
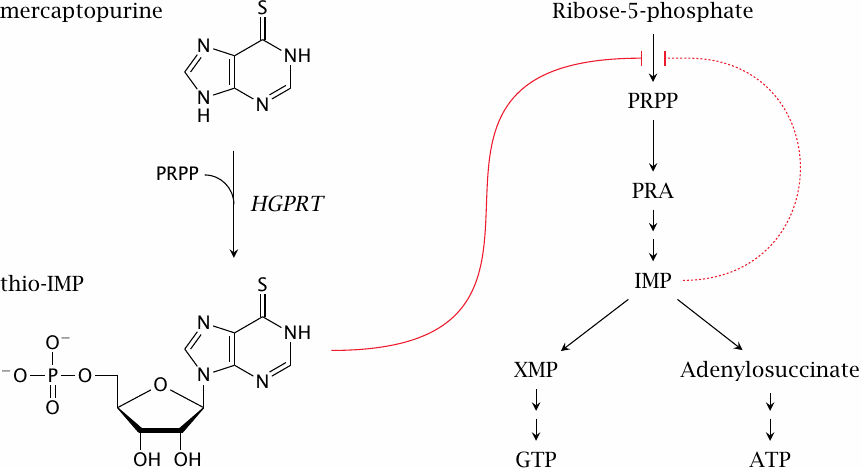
Mercaptopurine is activated to a nucleotide analogue by HGPRT, which also salvages the endogenous bases hypoxanthine and guanine (see slide 16.5.6). The analogue is not steered efficiently toward DNA incorporation. Instead, its main effect consists in the inhibition of de novo purine synthesis, by way of its similarity to inosine monophosphate, which inhibits the first enzyme in the pathway (ribose phosphate diphosphokinase). The related drug thioguanine acts in a similar manner. A low degree of DNA incorporation means low mutagenicity; like methotrexate, the two drugs are used not only in cancer but also in immunosuppressive therapy.
Mercaptopurine and thioguanine are metabolized through methylation by thiopurine methyltransferase, which uses S-adenosylmethionine as a cosubstrate. A defect of that enzyme, as such, has no manifest symptoms, but when treated with either drug these individuals may suffer severe myelosuppression, that is, disrupted cell proliferation in the bone marrow. It is therefore advisable, and in some cases mandatory, to test patients’ enzyme activities before using these drugs.
Mercaptopurine is also metabolized by xanthine dehydrogenase/oxidase. Combination of mercaptopurine with xanthine oxidase inhibitors such as allopurinol can again result in bone marrow toxicity, but is sometimes used deliberately to increase the immunosuppressive effect.
| 16.9.7 |
Structure of cytosine arabinoside (araC) and gemcitabine |
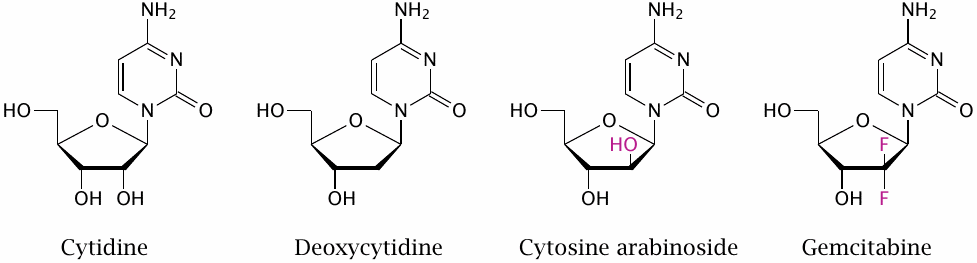
Cytosine arabinoside (cytarabine, araC) deviates from the normal cytosine nucleotides not in the base but in its sugar moiety, arabinose, which is epimeric to ribose at the second carbon atom. Like ribonucleosides, cytosine arabinoside can pass across the intestinal mucosa with the help of nucleoside transporters, which facilitates its uptake. Gemcitabine represents another variation on the same theme.
| 16.9.8 |
Metabolic activation and inactivation of araC |
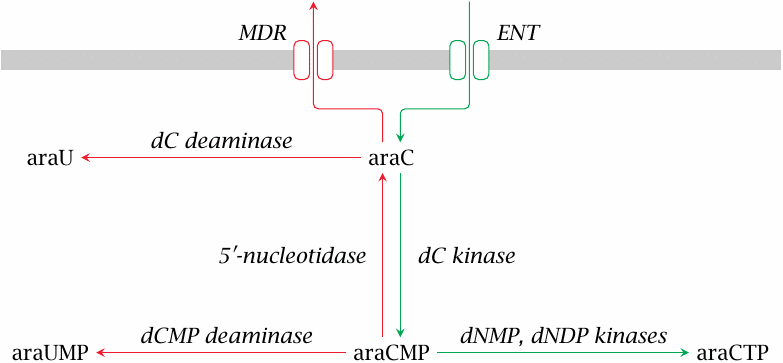
AraC enters the target cell by facilitated diffusion through the equilibrative nucleoside transporter (ENT). In order to become a substrate for DNA polymerase, araC must be phosphorylated to the triphosphate (araCTP); this is accomplished by several nucleoside and nucleotide kinases.
The activation of araC to araCTP can be intercepted at several stages. The extrusion of araC itself from the cell is mediated by multi-drug resistance transporters (MDR) such as P-glycoprotein, which belong to the family of ABC transporters (see slide 11.4.5). Like cytidine and deoxycytidine, araC can undergo deamination either as a free nucleoside or as a monophosphate, and the initial phosphorylation can be undone by the enzyme 5′-nucleotidase. Increased expression of MDR or of enzymes that counteract the activation of araC to araCTP cause tumor cell resistance (see slide 16.9.9).
| 16.9.9 |
Overexpression of 5′-nucleotidase in leukemic cells shortens survival |
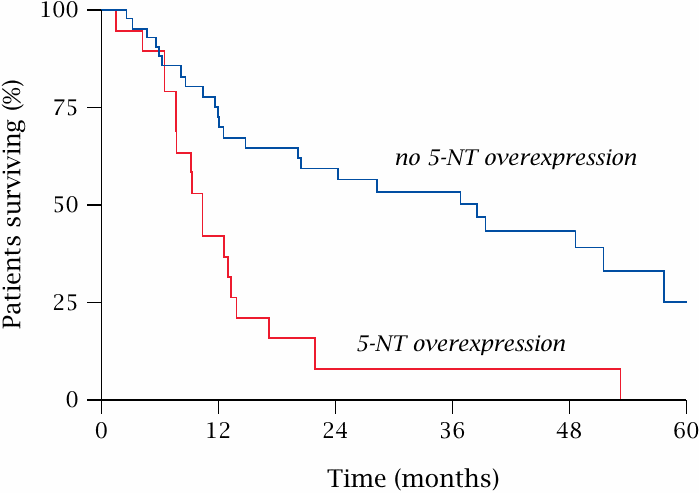
AraC is an important drug in the treatment of acute myeloic leukemia (AML). This plot shows that reduced susceptibility to araC due to increased expression of 5′-nucleotidase correlates with a significantly reduced duration of relapse-free survival in AML patients. Figure prepared from original data in [130].
| 16.9.10 |
Action mode of araCTP |
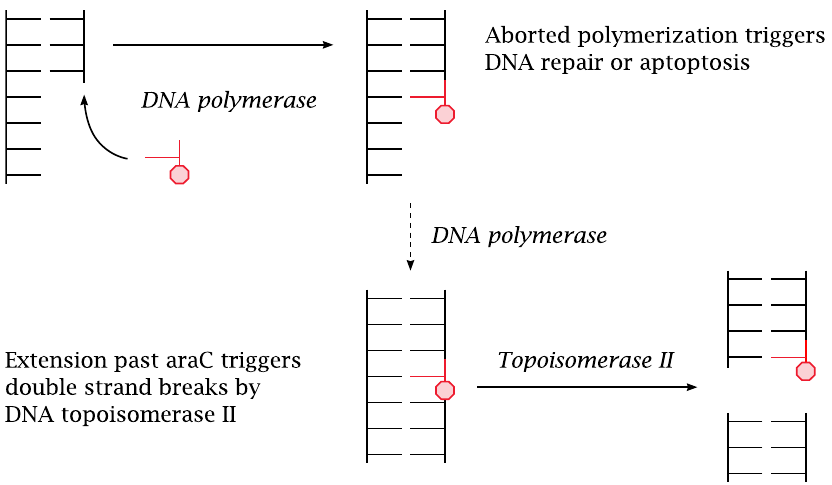
AraCTP is accepted as a substrate by DNA polymerase and becomes incorporated into a growing DNA strand. The polymerase may or may not manage to continue past an incorporated araC molecule. If it does not, DNA repair will be activated, and the base will be excised and replaced; this amounts to a delay of DNA synthesis and constitutes a proapoptotic signal.
If DNA polymerase does manage to continue past an incorporated araC molecule, the latter becomes part of a continuous DNA double strand. Such interposed araC residues then become preferred substrate sites for DNA topoisomerase II, which cleaves the DNA double strand [131]. Resealing of the double strand may fail and lead to chromosome breaks. If multiple chromosomes have been cleaved at the same time, the free ends may be joined the wrong way, which will result in chromosome translocations.
| 16.9.11 |
Dideoxyadenosine inhibits retroviral DNA polymerase |
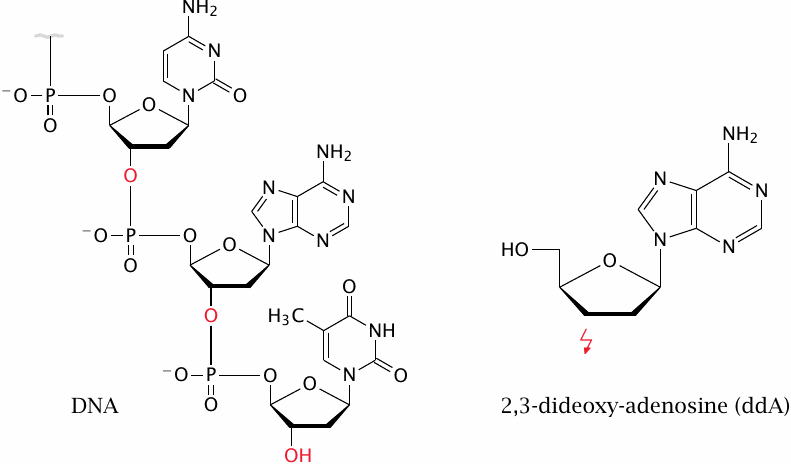
Base and nucleoside analogues are also useful in the treatment of viral diseases. An example is dideoxyadenosine (didanosine, ddA), which lacks the 3-OH group of the deoxyribose moiety. Since the 3- and the 5-OH groups form the bonds within the DNA strand, incorporation of ddA into a growing DNA strand causes termination of synthesis.101
The cellular DNA polymerases do not efficiently incorporate ddA, so it has limited cytotoxicity. However, the reverse transcriptase enzymes in retroviruses such as HIV use ddA efficiently, which leads to disruption of viral DNA synthesis. Inhibitors of reverse transcriptase such as ddA are a standard component of HIV therapy.
As stated above (slide 16.6.3), long-term application of didanosine may promote the manifestation of gout. Dideoxy-analogues of pyrimidines avoid this problem.
| 16.9.12 |
Aciclovir and ganciclovir |
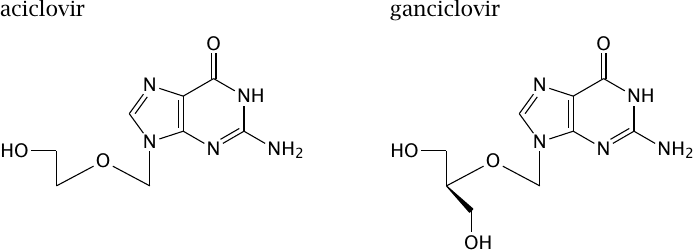
Aciclovir (or acyclovir) and ganciclovir are even more severely crippled than is dideoxyadenosine, and it is quite remarkable that some enzymes even accept them as substrates. This is the case with enzymes of viruses from the Herpes group, which includes herpes simplex virus, cytomegalovirus, varicella zoster virus, and a bunch of others. These are relatively large and complex viruses, which contain not only the bare minimum of enzymes such as nucleic acid polymerases but also their own nucleotide kinases.
| 16.9.13 |
Aciclovir: mode of action on herpes virus |
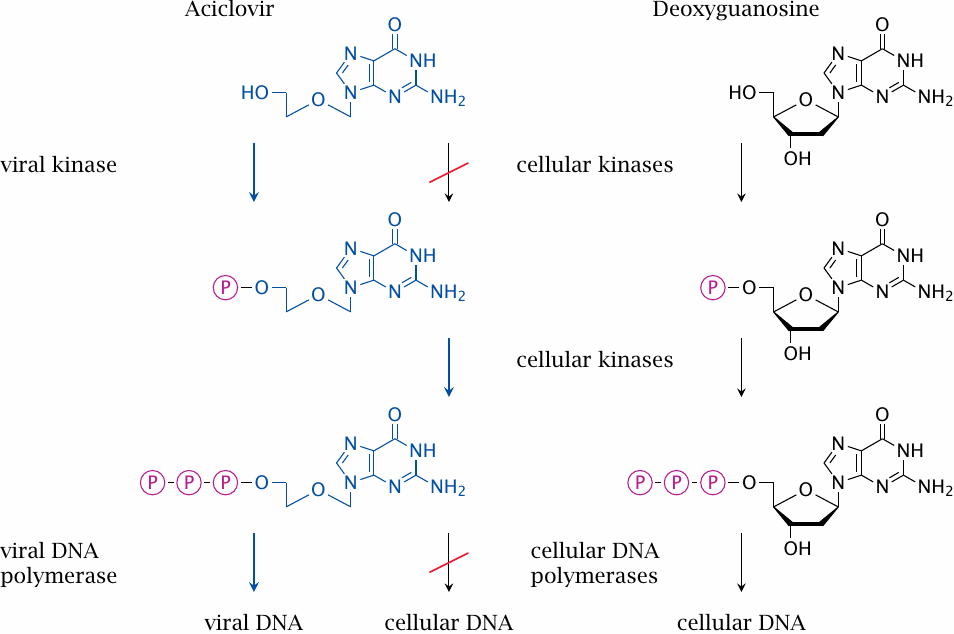
The selective toxicity of aciclovir for Herpes virus-infected cells, as opposed to normal cells, arises at two stages. Firstly, aciclovir is phosphorylated to the monophosphate only by viral nucleoside kinase, but not by the cellular enzyme. Secondly, the triphosphate, which is formed from the monophosphate by cellular kinases, is accepted as a substrate only by the viral DNA polymerase. This dual mechanism of selective toxicity causes aciclovir to be well tolerated yet effective.
| 16.9.14 |
Some more inhibitors of viral nucleic acid synthesis |
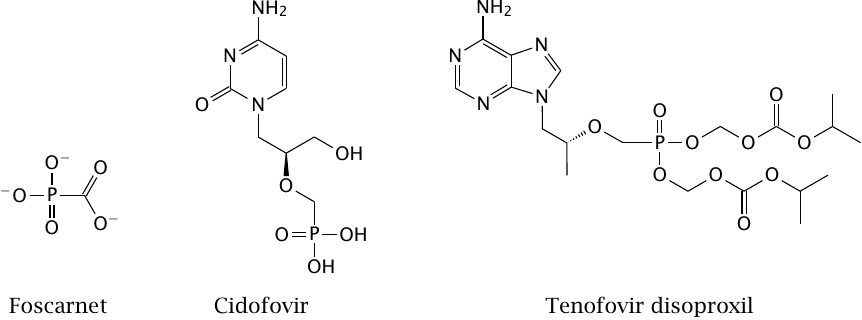
Foscarnet is an unusual drug. It is an analogue of pyrophosphate, which is released in each successive addition of a nucleotide to a growing DNA or RNA strand, and subsequently cleaved to phosphate:
| DNAn + dNTP | ⟶ | DNAn+1 + PPi | |
| PPi + H2O | ⟶ | 2 Pi |
Nucleic acid polymerases contain binding sites for the pyrophosphate (PPi) that is formed in the first step. Foscarnet lodges into the pyrophosphate binding sites of several viral polymerases. Unlike pyrophosphate, foscarnet is resistant to hydrolysis; it therefore remains bound to the polymerase enzymes and inhibits their activity.
Cidofovir is similar to aciclovir in having a mutilated ribose moiety. In addition, it also carries a phosphonate group; it therefore functionally resembles a nucleoside monophosphate and does not require the initial phosphorylation step, which gives it a broader spectrum than aciclovir. After addition of two more phosphates to this phosphonate group by cellular kinases, the drug inhibits several viral DNA polymerases.
Tenofovir disoproxil follows the same principle as cidofovir. However, it additionally carries two ester groups on the phosphonate. These groups make the drug more hydrophobic, which facilitates its diffusion across cell membranes. This improves the efficiency of intestinal uptake and distribution within the body. After cellular uptake, the ester groups are cleaved by esterases, which traps the molecule inside the cell and allows it to interact with nucleotide kinases and polymerases.