| 12 |
Amino acid metabolism |
| 12.1 |
Metabolic uses of amino acids |
- building blocks for protein synthesis
- precursors of nucleotides and heme
- source of energy
- neurotransmitters
- precursors of neurotransmitters and hormones
We have seen before that, during digestion in the gut, proteins are broken down to their constituent amino acids. Proteins contain twenty standard amino acids, which are incorporated into them during translation. Proteins may also contain several non-standard amino acids; these are mostly formed by post-translational modification, and they are much less abundant than the standard amino acids. In this chapter, we will focus on the standard amino acids, although some non-standard ones will appear in the urea cycle.
Animals and humans obtain many important metabolites from their food and have a fairly lazy synthetic metabolism. While many other organisms, even simple ones such as E. coli, can make all their amino acids from scratch, we possess synthetic pathways for only 11 out of the 20 standard amino acids. The remaining ones must be obtained from the diet and accordingly are referred to as the essential amino acids.75 A certain amount of dietary protein is therefore strictly necessary, and the lack of food protein is a very common form of malnutrition in impoverished countries. Nevertheless, during day-to-day protein turnover, most of the amino acids used in protein synthesis are obtained not from food but rather through endogenous protein breakdown; food protein only replaces the fraction of amino acids diverted toward other destinations.
In this chapter, we will mostly focus on degradative pathways of amino acids; their synthesis will be touched upon only briefly. The roles of specific amino acids in the synthesis of nucleotides are covered in chapter 16. The use of glycine in the synthesis of heme is discussed in chapter 17.
| 12.1.1 |
Outline of amino acid degradation |
- The liver is the major site of degradation for most amino acids, but muscle and kidney dominate the degradation of specific ones
- Nitrogen is removed from the carbon skeleton and transferred to α-ketoglutarate, which yields glutamate
- The carbon skeletons are converted to intermediates of the mainstream carbon oxidation pathways via specific adapter pathways
- Surplus nitrogen is removed from glutamate, incorporated into urea, and excreted
All amino acids contain at least one nitrogen atom, which forms their α-amino group; several amino acids contain additional nitrogen atoms in their side chains. Some nitrogen is used in biosynthesis, for example of nucleotides, but most of it is surplus and must be eliminated. To this end, the liver incorporates it into urea, which is released into the bloodstream and excreted by the kidneys.
Removal of nitrogen is typically an early step in the degradation and leaves behind the carbon skeleton. The structure of the latter is different for each amino acid, and accordingly each amino acid has its own specific pathway of degradation.
| 12.1.2 |
Amino acid breakdown pathways join mainstream carbon utilization at different points of entry |
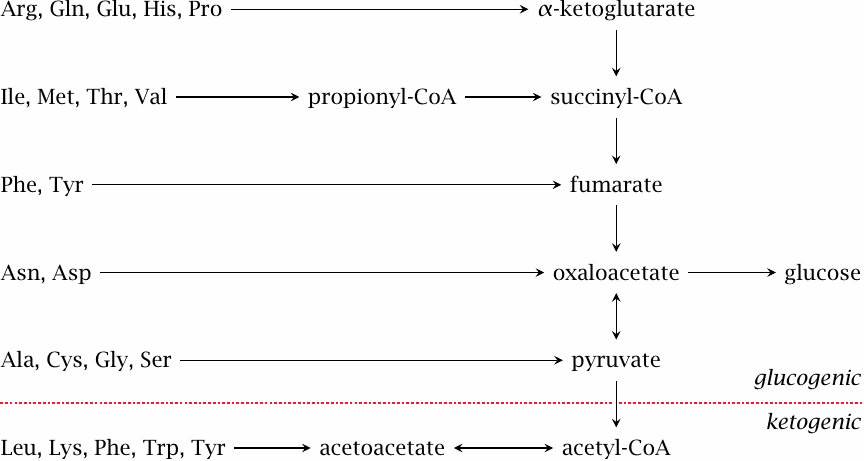
The degradative pathways can be divided into two major classes. As shown here, most amino acids are converted to intermediates of the citric acid cycle or to pyruvate, which in turn can serve as precursors for gluconeogenesis; these are the glucogenic amino acids. Those amino acids that yield acetoacetate are called ketogenic, since acetoacetate is one of the ketone bodies (see slide 10.4).
If you look carefully, you will see that phenylalanine and tyrosine are found on both sides of the divide, since they yield both fumarate and acetoacetate. I suppose that makes them both glucogenic and ketogenic, although some might insist that either category should take precedence; this is merely a matter of definition.
Depending on the composition of our diet, amino acids may be very important as a source of energy. While plant-derived foodstuffs are typically rich in starch, meat is high in protein but low in carbohydrates. Therefore, when on a diet that contains mostly meat, amino acids become our major source of glucose.
What happens if protein is supplied in excess of the amount needed to cover our requirement for glucose? While the liver in principle contains all enzyme activities required to oxidize the surplus carbon, measurements of the liver’s overall oxygen consumption indicate that most of the carbon must be disposed of elsewhere [81]. Presumably, much of the carbon is still initially converted to glucose, which is then either stored as glycogen or released into the circulation. In the latter case, the surplus will mostly be converted to fat in adipose tissue. A surplus of ketogenic amino acids will mostly be converted in the liver to triacylglycerol, which is then packaged and released as VLDL (see slide 11.4.2).
| 12.2 |
Transamination of amino acids |
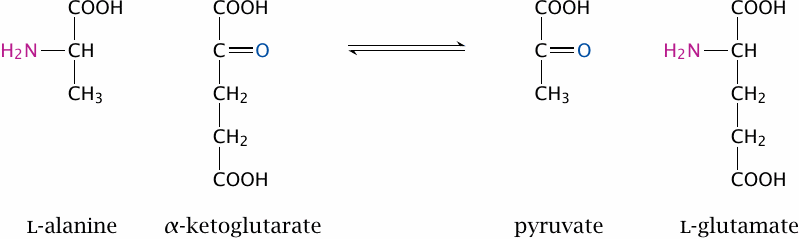
Most standard amino acids lose their α-amino group early on in degradation through transamination, that is, transfer to an α-keto acid. This is illustrated here for alanine, which transfers its amino group to α-ketoglutarate to become pyruvate.76 Transamination is mediated by several different aminotransferase enzymes. These may be specific for individual amino acids, or they may be able to process a group of chemically similar ones. The latter applies to the group of the branched-chain amino acids, which comprises leucine, isoleucine, and valine.
In the case of alanine, the α-keto acid that accepts the amino group is α-ketoglutarate; this also applies to most other amino acids. Transamination is freely reversible; therefore, both glutamate and α-ketoglutarate are substrates of multiple transaminases. If amino groups are to be transferred between two amino acids other than glutamate, this will usually involve the formation of glutamate as an intermediate. The role of glutamate in transamination is only one aspect of its central place in amino acid metabolism (see slide 12.3.7).
| 12.2.1 |
The reaction mechanism of transamination |
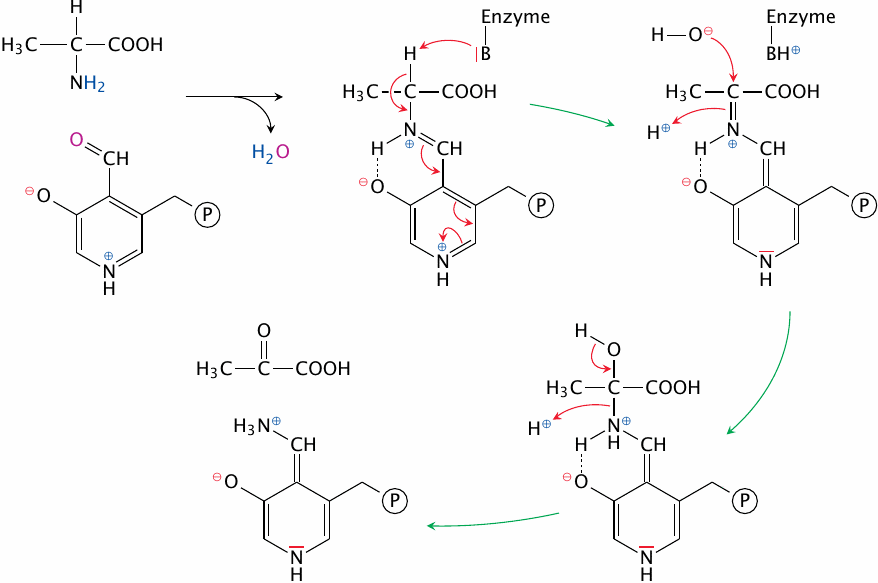
In this scheme, we again show alanine as the example substrate, but the mechanism is very general. All transaminases employ the coenzyme pyridoxal phosphate (PLP), which is the centerpiece of the reaction mechanism. The aldehyde group of PLP can form an aldimine, or Schiff base, with the α amino group of the substrate, which releases water.77
After binding of the substrate to PLP, a catalytic base in the active site abstracts its α hydrogen as a proton, and the surplus electron left behind travels all the way down to the nitrogen at the bottom of the PLP ring, inverting the entire sequence of single and double bonds along the way. This has the effect of turning the bond between the α carbon and the α nitrogen into a Schiff base, which then undergoes hydrolysis to release pyruvate.
At this point, the reaction is only half complete—the nitrogen is still attached to PLP and needs to be transferred to α-ketoglutarate. The sequence of steps involved in this transfer is the exact reversal of the ones shown here, so we won’t show them in detail. However, you might still find it a useful exercise to draw these reaction steps yourself.
It is interesting to compare the transaminase mechanism with that of serine hydroxymethyltransferase (slide 15.2.4). While that enzyme breaks the bond between the α carbon and the side chain rather than the amino group, it uses PLP in much the same manner to destabilize the substrate by withdrawing an electron. This also occurs in other PLP-mediated reactions; and in all of these reactions, PLP is often said to act as an electron sink. PLP also serves as a coenzyme in the glycogen phosphorylase reaction; however, its catalytic role there is entirely different (see slide 8.3.6).
| 12.2.2 |
The ping pong bi bi mechanism of transamination |
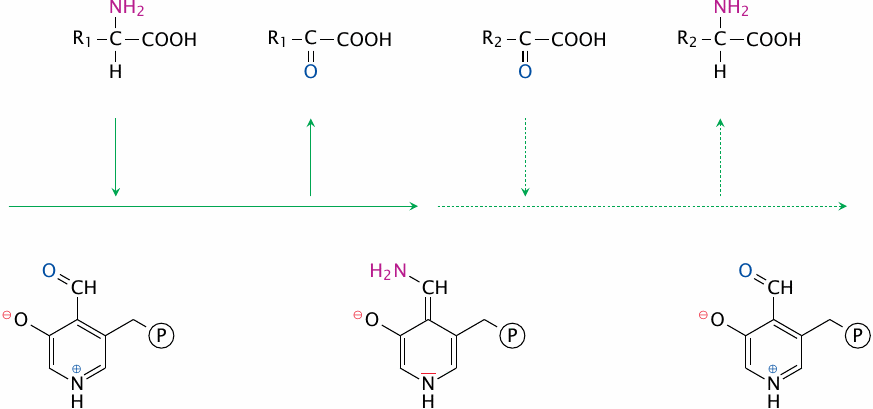
The interaction of the transaminase with its substrates involves four discrete steps: 1)the first substrate enters, 2)the first product leaves, 3)the second substrate enters, and 4)the second product leaves. Such a strictly sequential order is referred to as a ping pong mechanism. A ping pong bi bi mechanism is one that, on top of being strictly sequential, involves exactly two substrates and two products.78
While two different substrates must be used for the reaction to have some useful effect, it is of course possible for R1 and R2 to be identical—the reaction will work just fine, but simply achieve no net turnover.
| 12.3 |
Nitrogen disposal and excretion |
- Nitrogen accruing outside the liver is transported to the liver as glutamine or alanine
- In the liver, nitrogen is released as free ammonia
- Ammonia is incorporated into urea
- Urea is released from the liver into the bloodstream and excreted through the kidneys
While transamination solves the problem of removing the α-nitrogen for the amino acids other than glutamate, there also must be mechanisms for regenerating the α-ketoglutarate that is converted to glutamate in each transamination reaction, and for the ultimate disposal of nitrogen. A reaction that directly regenerates α-ketoglutarate is catalyzed by glutamate dehydrogenase, as follows:
| glutamate + H2O + NAD+ | ⟶ | α-ketoglutarate + NH3 + NADH + H+ |
While this reaction is straightforward, it produces free ammonia, which is quite toxic and must be kept at low (micromolar) concentrations in the systemic circulation at all times. Therefore, free ammonia is not a suitable medium for ultimate disposal of nitrogen; instead, elimination occurs mostly in the form of urea. The sequence of reactions that incorporates nitrogen into urea is the urea cycle.79
The glutamate dehydrogenase reaction is reversible in principle, but the affinity of the enzyme for ammonia is low. Interestingly, this enzyme can utilize both NAD+ and NADP+ as cosubstrates. As we have seen (slide 9.3.1), the former is present in the cell mostly in the oxidized form, which would favor the release of ammonia, whereas the latter is mostly found as NADPH, which would favor ammonia fixation. I have not been able to ascertain what regulatory mechanism, if any, prevents the enzyme from performing both reactions in a cycle, which would simply cause the reduction of NAD+ at the expense of NADPH.
| 12.3.1 |
The urea cycle, part 1: carbamoylphosphate synthetase |
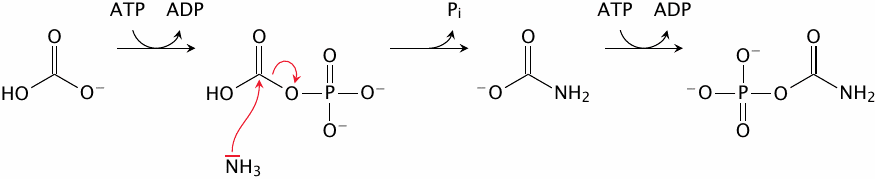
The urea cycle runs only in the liver. It begins with the incorporation of ammonia into carbamoylphosphate by the corresponding synthetase. This reaction occurs in three successive steps. The first step uses ATP to activate bicarbonate to carbonylphosphate, which then captures free ammonia to form carbamate. Another ATP-dependent step activates that intermediate to carbamoylphosphate. The carbamoyl group will find its way into the urea that is produced by the urea cycle.
| 12.3.2 |
The urea cycle, part 2: subsequent reactions |
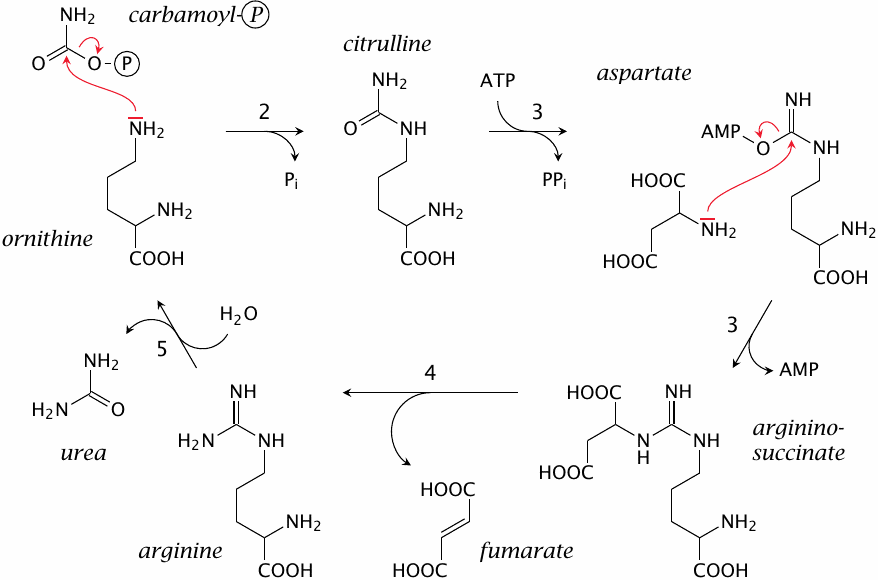
The subsequent reactions in the urea cycle are as follows:
- 2.The carbamoyl group is transferred from carbamoylphosphate to the δ-amino group of ornithine, a non-standard amino acid homologous to lysine, by ornithine transcarbamylase. This reaction yields citrulline.
- 3.Citrulline and aspartate form argininosuccinate, catalyzed by argininosuccinate synthetase. This reaction again requires ATP, which is converted to AMP in the process.
- 4.Argininosuccinate is cleaved to fumarate and arginine by argininosuccinase.
- 5.Urea is released from arginine by arginase, which regenerates ornithine and closes the cycle.
You will have noticed that only one of the nitrogens in urea is accounted for by carbamoylphosphate and, therefore, ammonia. The overall reaction of the urea cycle is
| NH3+HCO3–+aspartate | ⟶ | (NH2)2CO+fumarate |
with the additional expenditure of several equivalents of ATP in order to make things happen. Therefore, half of the nitrogen in urea is actually derived from aspartate, not ammonia. Where does this aspartate come from?
| 12.3.3 |
The urea cycle in context |
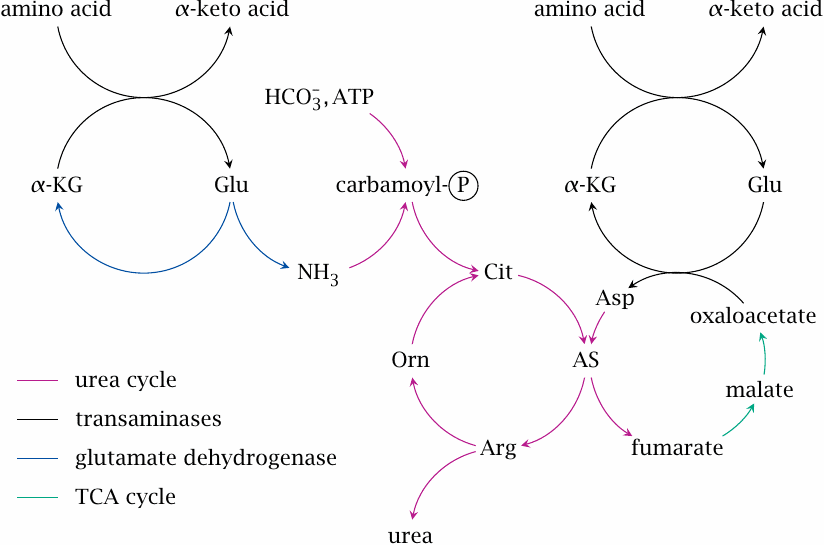
To answer this question, we just need to pull together our previous knowledge about transamination as well as the citric acid cycle. Fumarate is turned into malate and then oxaloacetate in the citric acid cycle, so we can just borrow those reactions. Oxaloacetate can be transaminated by aspartate aminotransferase using glutamate (slide 6.9.2), which in turn acquired its nitrogen by transamination of some other amino acid destined for degradation. In other words, the aspartate simply serves as an intermediate carrier of nitrogen en route from amino acid degradation to urea synthesis.
The network of reactions shown in this slide accounts for the disposal of nitrogen that accrues in amino acid degradation in the liver. As stated at the outset, other tissues also break down amino acids; for example, skeletal muscle metabolizes the lion’s share of the branched-chain amino acids. Therefore, a mechanism is needed to ferry the nitrogen produced in the peripheral organs to the liver. Ammonia cannot be used as a carrier, since it is too toxic; amino acids are a better alternative. The two most important nitrogen carriers are alanine and glutamine (see below).
| 12.3.4 |
The urea cycle spans mitochondria and cytosol |
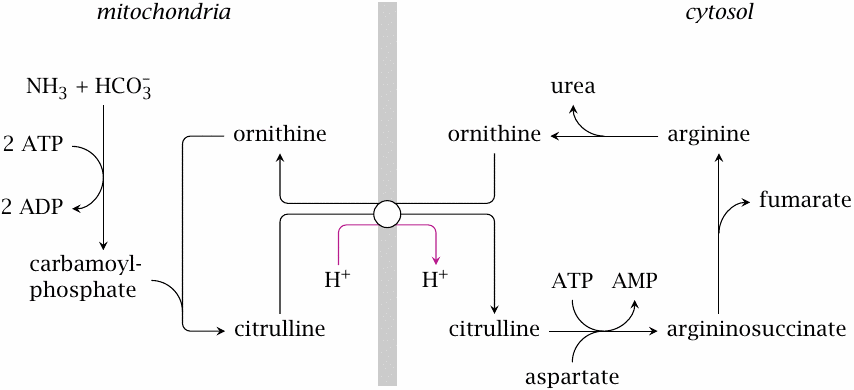
The carbamoylphosphate synthetase reaction and the ornithine transcarbamylase reaction occur inside the mitochondria, while the remaining reactions take place in the cytosol. This requires the transport of ornithine and citrulline, which are exchanged for each other by a specific transporter in the inner mitochondrial membrane.
Ornithine has two free amino groups, while citrulline has one. The exchange transport is rendered electrically neutral by the cotransport of a proton out of the mitochondria, that is, against its concentration gradient. The energetic cost of this uphill transport is offset by the expenditure of ATP in other steps of the urea cycle. Nevertheless, the coupling of this substrate exchange to proton export will keep the cytosolic concentration of citrulline low at equilibrium.
Also note that the reactions that involve fumarate and aspartate occur in the cytosol. We had just noted that the conversion of fumarate back to aspartate involves some reactions borrowed from the TCA cycle. That cycle runs in the mitochondria; however, fumarate does not need to enter the mitochondria at this stage, since all required enzyme activities are also present in the cytosol.
| 12.3.5 |
The glucose-alanine cycle |
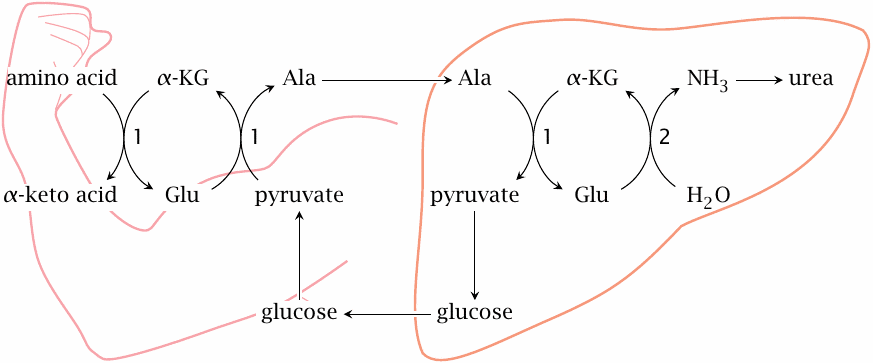
The glucose-alanine cycle is an interorgan cycle that piggybacks on the Cori cycle and accomplishes a net transport of nitrogen from muscle and other peripheral tissues to the liver. Here, pyruvate that is produced from glucose in the periphery is not reduced to lactate—as is the case in the Cori cycle, see slide 8.5.3—but instead transaminated to alanine, which is then transported to the liver. There, transamination is reversed, and pyruvate is converted again to glucose by gluconeogenesis. Release of glucose into the bloodstream and renewed glycolysis in the periphery close the cycle.
Apart from the reactions of glycolysis and gluconeogenesis, the cycle involves various transaminases (1) and glutamate dehydrogenase (2).
| 12.3.6 |
Nitrogen transport by glutamine |
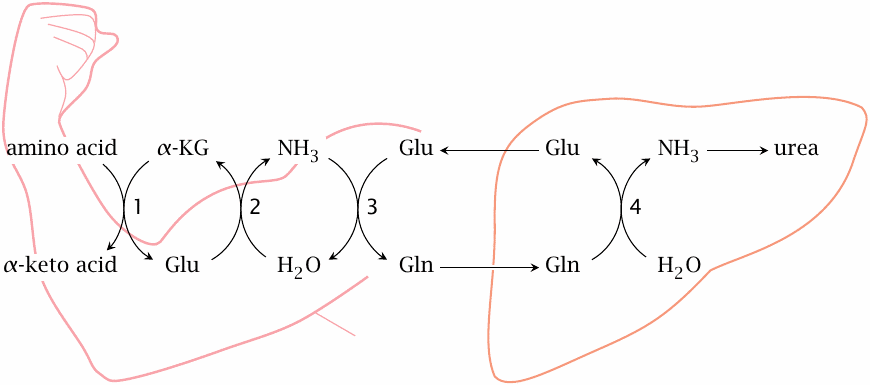
Glutamine is the most abundant amino acid in the blood; it is significant both as a nitrogen and a carbon carrier.80 It can bring about a net transfer of nitrogen from peripheral tissues to the liver in exchange for glutamate. The enzymes involved in the overall scheme are transaminases (1), glutamate dehydrogenase (2), glutamine synthetase (3), and glutaminase (4). The latter two reactions are shown in detail in slide 12.3.7.
One might reason that, in the liver, glutamate could be further deaminated by glutamate dehydrogenase, and α-ketoglutarate be returned to the periphery, which would allow the transfer of two nitrogen atoms in each turn of the cycle. This should work in principle, but the plasma concentration of α-ketoglutarate is too low for it to be quantitatively important.
| 12.3.7 |
The central role of glutamate in nitrogen disposal |
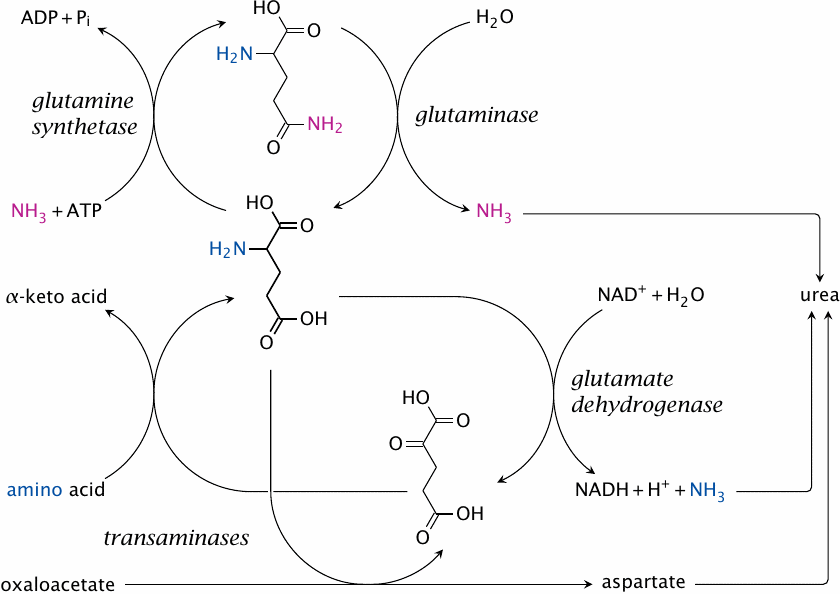
If we summarize the network of reactions in nitrogen elimination, we find that glutamate has a central place in it. Glutamate collects nitrogen from other amino acids through transamination and either releases it as ammonia or transfers it to aspartate in order to feed the urea cycle. Together with glutamine, it also controls the level of free ammonia and accomplishes the transport of nitrogen between organs.
As shown in this scheme, glutamate is formed from glutamine by glutaminase, and it can be turned back into glutamine by glutamine synthetase.81 Evidently, both enzymes together would create a futile cycle that would accomplish nothing except ATP hydrolysis. In most organs, only one or the other enzyme has significant activity; for example, glutamine synthetase predominates in skeletal muscle (see slide 12.3.6), whereas glutaminase is abundant in the kidneys, which use it to secrete ammonium chloride into the urine when eliminating excess acid.
The liver contains both glutaminase and glutamine synthetase, which would suggest that futile cycling should occur. However, as it turns out, the enzymes are present inside the same tissue but not the same cells. Instead, they are distributed strategically within the liver lobule so as to create a confined compartment to host the urea cycle (see next slide).
| 12.3.8 |
Control of ammonia levels in the liver lobule |
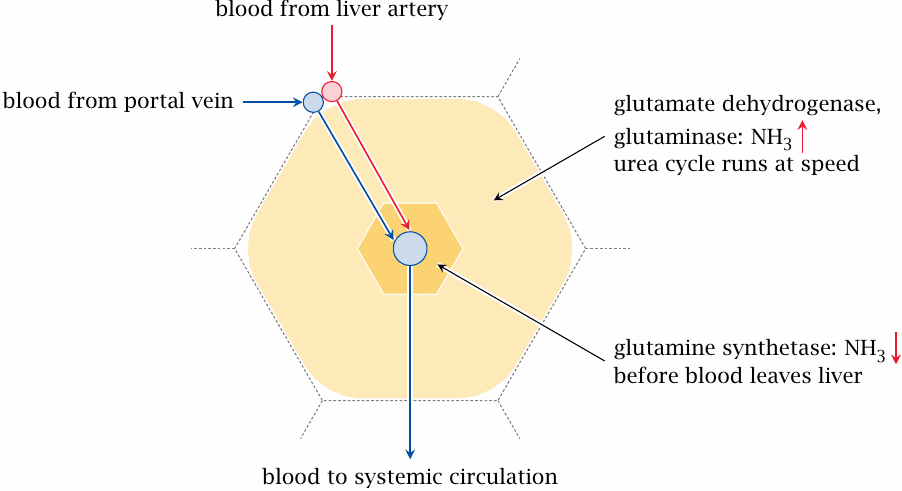
The toxicity of ammonia mandates that its concentration be kept very low in the systemic circulation. On the other hand, for the urea cycle to run at speed, the concentration must be high enough to saturate the initial enzyme, carbamoylphosphate synthetase, to a useful degree. Therefore, ammonia must be released when the blood enters the liver tissue, and scooped up again before the blood is drained away into the general circulation.
To make this work, the enzymes that release or fix ammonia, respectively, are strategically distributed in the liver tissue. We had seen before that the liver consists of functional units called lobules, with the blood filtering through each lobule from the periphery towards the center, from where it is drained toward the systemic circulation (slide 1.6.3). Glutaminase and glutamate dehydrogenase, which release ammonia, are found predominantly in the periphery of the lobule, the so-called periportal zone. Here, they increase the concentration of free ammonia, allowing the urea cycle to run at speed. As the blood seeps into the pericentral zone that surrounds the lobule’s central vein, ammonia is scavenged again by glutamine synthetase before the blood is drained from the liver into the general circulation.
A similar spatial separation applies to the enzymes of arginine degradation. The first step in this pathway is catalyzed by arginase, which also functions in the urea cycle and produces ornithine. In the periportal zone, it would be deleterious to continue the degradation beyond ornithine, since this would drain the urea cycle of its intermediates. Therefore, the next enzyme in the pathway, ornithine aminotransferase, is only found in the pericentral zone, in which the urea cycle must shut down anyway.
The purposeful distribution of different enzyme activities illustrates nicely how biochemical and anatomical levels of organization are interrelated, and how our body is not just a bag of cells, not even at the level of individual organs and tissues.
| 12.3.9 |
Regulation of the urea cycle |
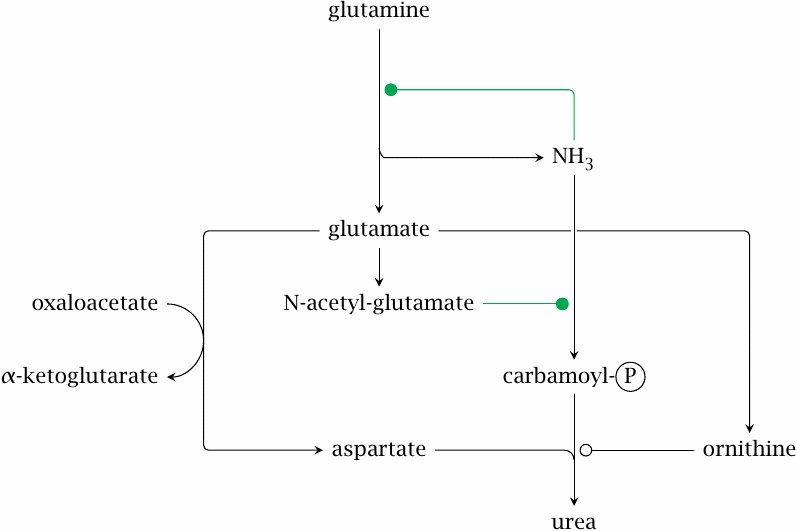
At the heart of the mechanisms that control flow through the urea cycle, we once again find glutamine and glutamate. Glutaminase gets the ball rolling by releasing ammonia from glutamine. Quite unusually, ammonia exercises positive rather than negative feedback on glutaminase, which causes a rapid accumulation of both glutamate and ammonia. High levels of glutamate then promote the incorporation of ammonia into urea in several ways.
- 1.Some glutamate is converted to N-acetylglutamate. This is a regulatory molecule that allosterically activates carbamoylphosphate synthetase.
- 2.Glutamate is also the biosynthetic precursor of ornithine. A high level of glutamate will also raise the level of ornithine, which will increase the flow through the urea cycle.
- 3.As we have seen, glutamate itself feeds nitrogen into urea synthesis via glutamate dehydrogenase or via aspartate.
Note that all these regulatory events amplify the flow through the urea cycle; they are the ones that run in the periportal zone of the liver lobule. As stated above, the urea cycle is shut down in the pericentral zone through the capture of remaining ammonia by glutamine synthetase as well as by ornithine degradation.
| 12.3.10 |
Hereditary enzyme defects in the urea cycle |
- may affect any of the enzymes in the cycle
- urea cannot be synthesized, nitrogen disposal is disrupted
- ammonia accumulates, as do other metabolites depending on the deficient enzyme
-
treatment
- protein-limited diet
- arginine substitution
- alternate pathway therapy
Urea cycle defects primarily become symptomatic due to the accumulation of ammonia, which impairs brain function. Another aspect is the deficiency of arginine. In healthy individuals, arginine can be diverted from the urea cycle toward protein synthesis as required, but this supply is lacking if the cycle is disrupted. This may induce protein catabolism, thereby exacerbating the symptoms. The problem is addressed by the addition of arginine to the diet. If the enzyme defect is not between citrulline and arginine, it is possible to supply citrulline instead; this has the advantage of picking up one nitrogen equivalent en route to arginine.
The rationale for treating these enzyme defects with a protein-restricted diet is fairly obvious. Another, more intriguing approach is known as alternate pathway therapy. Here, the patients are given several innocuous organic acids that are substrates for conjugation with amino acids. These conjugates then serve as an alternative vehicle for the renal elimination of surplus nitrogen. This ingenious form of treatment is further discussed in slide 19.3.8.
| 12.4 |
Degradative pathways of individual amino acids |
The degradation pathways for the individual amino acids vary considerably in complexity. As we had seen, some amino acids only require a single transamination step; on the other hand, others have lengthy degradation pathways with intriguing catalytic mechanisms. We will here consider some selected examples; several others are discussed in a later chapter (slides 15.2.4–15.2.7).
| 12.4.1 |
Asparagine degradation |
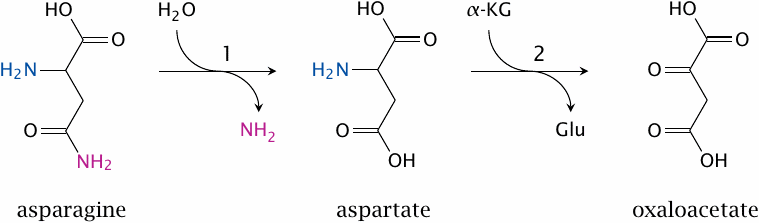
Asparagine is homologous to glutamine, and its degradation is analogous, too—just like glutaminase produces glutamate, asparaginase (1) produces aspartate, which can then be transaminated by aspartate aminotransferase (2) to oxaloacetate. Asparaginase is mentioned here not for any notable chemistry, but rather because of some interesting medical context.
Asparagine is a non-essential amino acid, which means that it can be synthesized by human cells; the enzyme responsible for this, asparagine synthetase, uses glutamine as its amide group donor. Nevertheless, in some forms of leukemia, the leukemic cells lack the synthetic capacity for asparagine. This can be exploited for therapy—the leukemia patients are treated with intravenous application of asparaginase.82 This lowers the serum level of asparagine and therefore starves the leukemic cells.
| 12.4.2 |
Serine dehydratase |
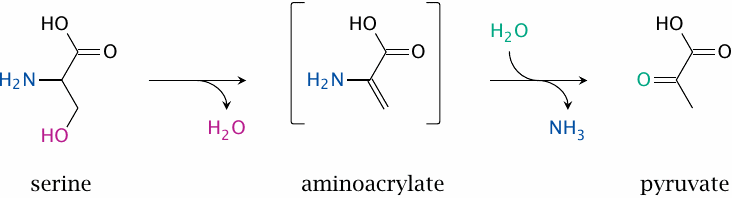
Serine, another non-essential amino acid, can be degraded along several different pathways; this slide shows one of them. Only the first step is enzymatically catalyzed; the aminoacrylate produced is unstable and spontaneously hydrolyzes to pyruvate. The second step releases ammonia, which must be disposed of. It seems that in humans the reaction occurs only in the liver, where the ammonia can directly enter the urea cycle.
Like the transaminases, the enzyme uses pyridoxal phosphate, and the role of the coenzyme is often presented along the lines of the usual electron sink mechanism (see slide 12.2.1). However, based on the crystal structure of the enzyme, a different mechanism has been proposed, in which no electron sink appears and instead the phosphate group of PLP plays a prominent role [82]. I am not enough of a chemist to judge how plausible this mechanism may be.
| 12.4.3 |
Serine-pyruvate transaminase |
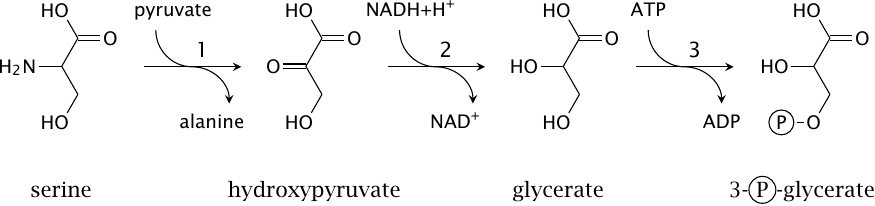
An alternative pathway starts with the transamination of serine rather than with deamination. In a departure from the usual routine, the transaminase employs pyruvate rather than α-ketoglutarate as its other substrate, which yields alanine (1); subsequently, of course, the nitrogen may yet be transferred from alanine to α-ketoglutarate in a second transamination. Serine itself is converted to hydroxypyruvate, which is then reduced to glycerate by hydroxypyruvate reductase (2). Glycerate kinase (3) produces 3-phosphoglycerate.
While both pyruvate, which is produced by serine dehydratase, and the 3-phosphoglycerate produced here can serve as substrates for gluconeogenesis, the transamination pathway shown in this slide avoids the release of free ammonia. It would therefore be preferable in tissues other than the liver.
A third alternative for serine degradation is provided by serine hydroxymethyltransferase, which produces N,N’-methylene-tetrahydrofolate and glycine. This pathway is shown in slide 15.2.4.
| 12.4.4 |
Degradation of leucine |
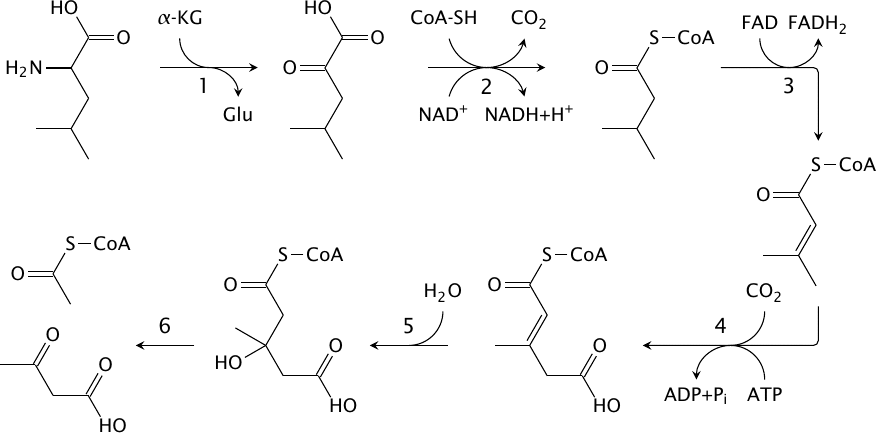
Leucine, isoleucine and valine are collectively referred to as the branched-chain amino acids. Unlike the other amino acids, these ones undergo degradation mostly in skeletal muscle. This is reminiscent of fatty acids, which are also degraded prominently in muscle, and indeed several steps in leucine degradation have similarity with the reactions we have seen in fatty acid metabolism. Leucine degradation involves the following steps:
- 1.Transamination by branched chain amino acid (BCAA) transaminase yields α-ketoisocaproate.
- 2.α-Ketoisocaproate is decarboxylated and dehydrogenated by branched chain α-keto acid dehydrogenase. Like the transaminase in step 1, this dehydrogenase participates in the degradation of all branched chain amino acids (valine, leucine, isoleucine). The reaction mechanisms and the structural organization of this enzyme are completely analogous to pyruvate dehydrogenase and α-ketoglutarate dehydrogenase, and all use the very same E3 subunit (see slide 5.4.4).
- 3.The resulting metabolite, isovaleryl-CoA, is similar in structure to a fatty acyl-CoA. It likewise undergoes a FAD-dependent dehydrogenation reaction (by isovaleryl-CoA dehydrogenase), which yields isopentenyl-CoA.
- 4.Biotin-dependent carboxylation yields methylglutaconyl-CoA. In all of the previous carboxylation reactions we have seen, carboxylation was facilitated by a vicinal carbonyl group, which tends to withdraw electrons from the carbon being carboxylated. This electron-withdrawing effect can be relayed by a conjugated C=C double bond, which is the case here.83
- 5.Addition of water by methylglutaconyl-CoA hydratase yields HMG-CoA.
- 6.HMG-CoA is split by HMG-CoA lyase to acetyl-CoA and acetoacetate.
The final reaction occurs in the very same way in ketone body synthesis (see slide 10.4.1). Evidently, therefore, leucine is a purely ketogenic amino acid.
| 12.4.5 |
Degradation of phenylalanine and tyrosine |
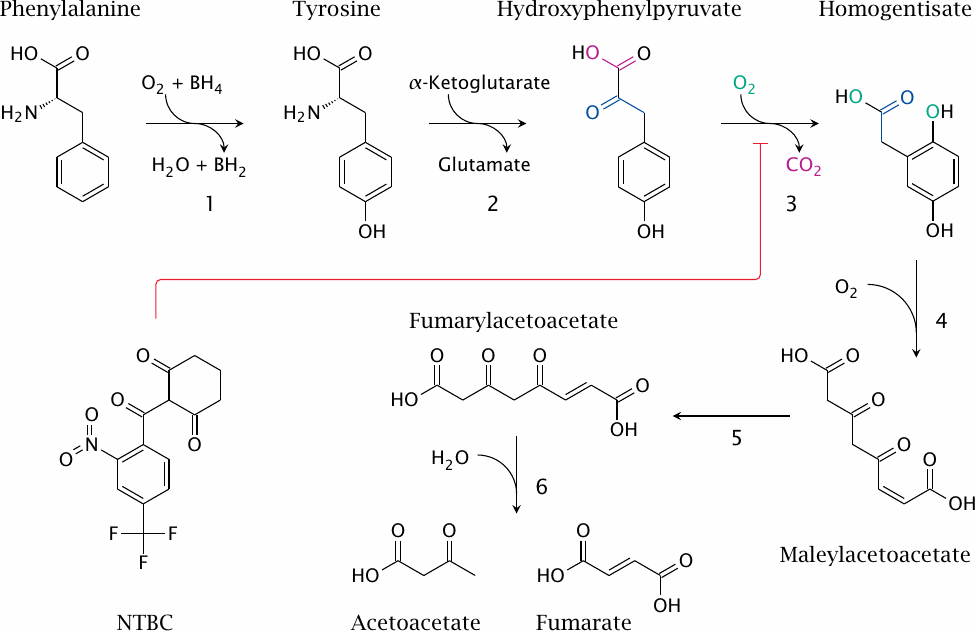
While the degradation of leucine shows a comforting similarity to previously encountered pathways, the situation is profoundly different with phenylalanine. This is due to the aromatic nature of the side chain. Aromatic rings are quite stable, and therefore some brute force is needed to crack them open. The best tool for this task is molecular oxygen, and a liberal dose of it is used in the breakdown of phenylalanine. The pathway involves the following reactions:
- 1.Phenylalanine is converted to tyrosine by phenylalanine hydroxylase. In this reaction, the second oxygen atom is released as water, reduced at the expense of the redox cosubstrate tetrahydrobiopterin (BH4).
- 2.Tyrosine transaminase yields p-hydroxyphenylpyruvate.
- 3.p-Hydroxyphenylpyruvate dioxygenase uses another oxygen molecule to turn hydroxyphenylpyruvate into homogentisate. I have traced the bits and pieces in this reaction using colors, but an actual explanation of the advanced magic performed by the enzyme is beyond my feeble powers of narration.
- 4.Ring cleavage by homogentisate dioxygenase uses a third molecule of oxygen. The product is maleylacetoacetate.
- 5.Maleylacetoacetate isomerase produces fumarylacetoacetate.
- 6.Fumarylacetoacetate hydrolase releases fumarate and acetoacetate.
Since the first transformation of phenylalanine yields tyrosine, it follows that the pathway accounts for the degradation of both amino acids, and moreover that tyrosine is not an essential amino acid. Of the two final products, fumarate can enter gluconeogenesis, while acetoacetate cannot.
The drug 2-[2-Nitro-4-(trifluoromethyl)benzoyl]cyclohexane-1,3-dione84 (NTBC), also shown in this slide, is an inhibitor of p-hydroxypyruvate dioxygenase. The use of this inhibitor in tyrosinemia will be explained below.
| 12.5 |
Hereditary enzyme defects in amino acid metabolism |
Since there are so many different pathways for the degradation of the various amino acids, it is understandable that many of the known inborn errors of metabolism are related to amino acid metabolism. We will consider a few examples that affect the pathways discussed here.
| 12.5.1 |
Phenylketonuria (PKU) |
- homozygous defect of phenylalanine hydroxylase
- affects one in 10,000 newborns among Caucasians; frequency differs with race
- excess of phenylalanine causes symptoms only after birth; intrauterine development normal
- cognitive and neurological deficits, probably due to cerebral serotonin deficit
- treatment with phenylalanine-restricted diet
- some cases are due to reduced affinity of enzyme for cofactor THB, can be treated with high dosages of THB
As with most genetic enzyme defects, the clinical disease is manifest only in homozygous individuals. Dietary phenylalanine that is not used for protein synthesis accumulates and causes toxicity. It appears that the excess phenylalanine crowds out tryptophan at the l-aromatic amino acid transporter in brain capillaries. This transporter keeps the brain supplied with all aromatic amino acids. Since tryptophan is the precursor of the neurotransmitter serotonin, the competitive inhibition of its transport to the brain results in a lack of cerebral serotonin [83], which is believed to cause the observed deficits in brain function and development.
In addition to phenylalanine itself, some aberrant metabolites derived from it also occur at increased levels, and the appearance of ketone derivatives such as phenylpyruvic acid in the urine has given the disease its name. These metabolites have no proven connection to the pathogenesis of the disease.
| 12.5.2 |
The Guthrie test for diagnosing phenylketonuria |
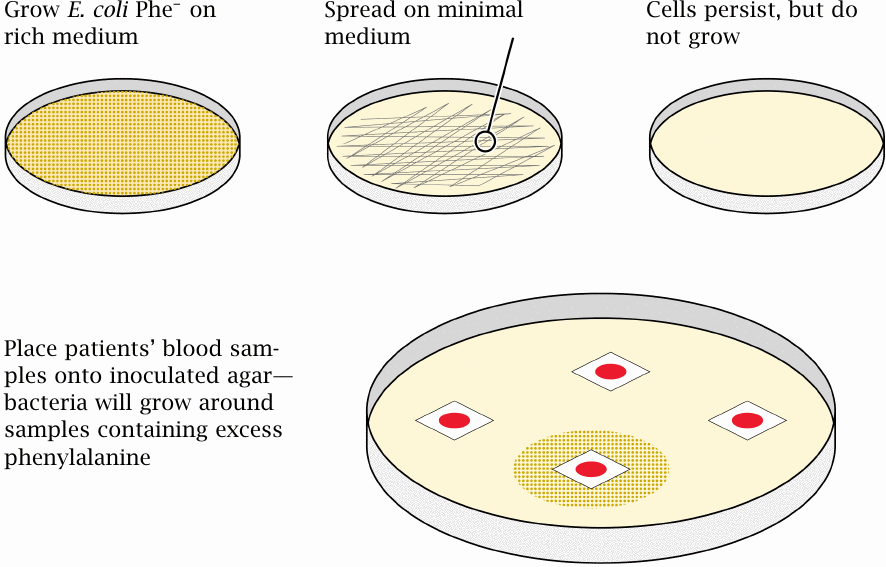
The treatment of phenylketonuria is pretty straightforward: Limitation of dietary phenylalanine. Tyrosine is sufficiently available in a reasonably protein-rich diet, so that the lack of its endogenous formation won’t be a problem. The challenge, then, is to diagnose the disease in newborn kids, before any damage is done. Happily, the enzyme defect does not cause a problem during fetal development, since the placenta constantly equilibrates both useful and potentially harmful metabolites between the maternal and the fetal circulation. Buildup of a metabolite in the fetus will therefore not occur as long as the mother’s metabolism is able to degrade it.
The modern test for phenylketonuria is effective but boring—a sample of blood is drawn, and the phenylalanine concentration in the serum is determined by HPLC. The original test—the Guthrie test—was a bit more roundabout in principle, yet ingenious and exceedingly simple and cheap in practice. Moreover, it well illustrates the power of bacterial genetics in biochemistry, and it therefore merits discussion here.
In contrast to mammalian cells, the bacterium Escherichia coli can synthesize all 20 standard amino acids, as long as it has ammonia, some inorganic salts, and an organic carbon source such as glucose. Such a substrate mixture constitutes a minimal medium. The Guthrie test makes use of a mutant E. coli strain that is Phe−, which means that it is unable to synthesize phenylalanine on its own. This strain can be grown on a rich medium that supplies phenylalanine; however, when spread onto minimal medium, it will not grow.
Now, if we take a little snippet of filter paper soaked with a drop of baby blood and place it on top of the inoculated minimal medium, any phenylalanine contained in it will diffuse into the surrounding agar. If there is enough of it in the sample, this will allow the bacteria in the vicinity to resume growth. Therefore, a zone of bacterial growth surrounding a blood sample will identify a patient with phenylketonuria. Brilliant!
Note that, for this test to work, we cannot collect the blood sample right away after birth. As noted above, the fetal blood equilibrates with the mother’s, and so the phenylalanine concentration in the blood of a newborn with the disease is only slightly increased at birth. We therefore must allow 1–2 weeks after delivery for phenylalanine to accumulate in the child’s blood for the Guthrie test to respond. This is a drawback of the test relative to the HPLC method—the latter is more quantitatively accurate and readily detects the smaller increase in phenylalanine concentration that is present at the time of delivery.
| 12.5.3 |
Ochratoxin A inhibits phenylalanyl-tRNA synthetase |
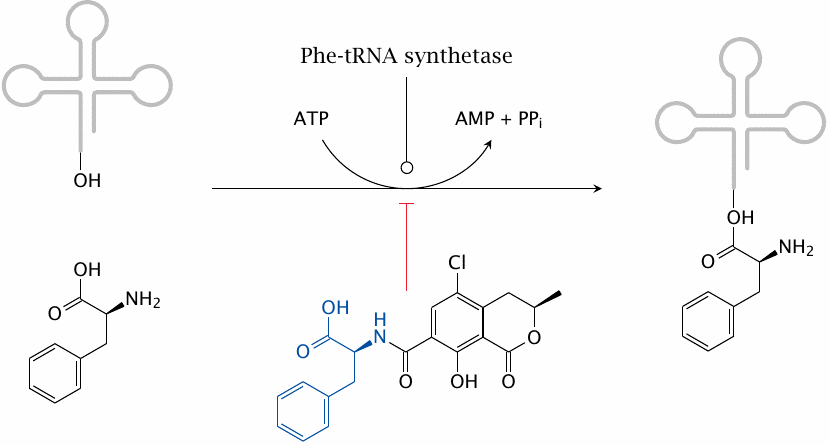
The variation of the gene frequency for PKU between races and geographical areas suggests that some regional environmental conditions may confer a selective advantage to the heterozygous state, as is the case with sickle cell anemia and glucose-6-phosphate dehydrogenase deficiency in regions with endemic malaria. It has been proposed that the heterozygote advantage in PKU consists in protection from the fungal toxin ochratoxin A, which is produced by some Aspergillus molds that cause food to rot [84].
Ochratoxin A competitively inhibits the coupling of phenylalanine to its cognate tRNA by the corresponding aminoacyl transferase and thereby disrupts protein synthesis. It is more toxic to fetuses than to adults, most likely because fetuses are short of the enzymes that inactivate xenobiotics and toxins such as ochratoxin. Mothers who are heterozygous for PKU will have a somewhat higher level of phenylalanine, which will be shared with the fetus via the placenta. This will counter the inhibition of tRNA aminoacylation in the fetus and thereby afford it some measure of protection.
One of the places with the highest abundance of PKU is Ireland. This country is also known for its repeated historic episodes of severe famine. Starving people will tend to eat rotten food rather than discard it. Indeed, reference [84] reports that, among Irish women, lower rates of abortion were found in those who were heterozygous for PKU. I have not ascertained whether the time periods covered by those statistics coincided with periods of actual famine.
| 12.5.4 |
Tyrosinemia |
- homozygous defect of fumarylacetoacetate hydrolase
- fumarylacetoacetate and preceding metabolites back up
- fumaryl- and maleylacetoacetate react with glutathione and other nucleophiles, causing liver toxicity
- the drug NTCB inhibits p-hydroxyphenylpyruvate dioxygenase, intercepting the degradative pathway upstream of the toxic metabolites
- dietary restriction of tyrosine required to prevent neurological deficit
Tyrosinemia is comparatively common in Quebec. In this case, there seems to be no heterozygote advantage; instead, the high incidence is due to the so-called founder effect, that is, the common descent of the afflicted population from a small group of founding settlers that happened to contain one or several carriers of the gene. (Refer to slide 12.4.5 for the relevant pathway and enzyme reactions.)