| 20 |
Enzyme and gene therapy of enzyme defects |
| 20.1 |
General considerations |
- How many organs are affected by the enzyme defect: One organ, a few, or all organs?
- How severe is the defect?
- Can the defect be adequately controlled by conventional treatment?
While some enzymes, such as pyruvate dehydrogenase, are expressed and required in almost all cells, others are expressed exclusively or preferentially in selected organs. For example, UDP-glucuronosyltransferases are expressed mostly in the liver, and enzyme defects accordingly affect this organ the most.
The clinical severity of enzyme defects ranges from benign to fatal. A fairly benign defect is the Gilbert syndrome, in which bilirubin glucuronidation proceeds at a reduced rate. This condition requires nothing more than caution in the selection and dosage of drugs when treating other diseases. On the other hand, untreated adenosine deaminase deficiency is fatal (see sections 16.5 above and 20.2 below). Obviously, aggressive therapeutic strategies such as organ transplants or gene therapy are warranted only with severe defects.
| 20.1.1 |
Conventional therapeutic strategies |
- diets
- drugs
- organ transplants
Diets can be used in many enzyme defects, and if effective, they are certainly to be preferred. An example is the fructose- and sucrose-free diet in fructose intolerance, which effectively prevents the deleterious depletion of free phosphate and of ATP in this disease (see slide 4.2.2).
Drugs are useful in several hereditary enzyme defects, but usually in a limited way. We have already discussed the use of NTCB in tyrosinemia (slide 12.5.4) and of organic acids in urea cycle defects (slide 12.3.10); a few more examples are discussed below. Organ transplants are useful in those diseases that primarily affect a single organ, typically the bone marrow or the liver.
| 20.1.2 |
Therapeutic strategies based on molecular biology |
Correction of …
- DNA: gene therapy
- mRNA: suppression of mutant stop codons with drugs
- protein: enzyme substitution
Enzyme defects are in principle the most logical and suitable targets for gene therapy. A key advantage of gene therapy is its long-lasting effect—at least in principle, the treatment maybe be effective for life. However, in most enzyme defects, gene therapy is still at the experimental stage, and it has not yet become the standard therapy in any specific disease.
Enzyme substitution therapy is more widely used. Since the enzyme molecules have a limited half-life, the enzyme has to be applied regularly, often in weekly or bi-weekly intervals. While this is costly and somewhat inconvenient, it has the advantage that the therapy can simply be discontinued in case of untoward reactions, which would of course not be feasible with gene therapy.
Correction at the mRNA level through suppression of stop codons—also referred to as translational antitermination—is confined to a few specific examples, since most gene defects that inactivate an enzyme do not involve mutant stop codons.
| 20.1.3 |
Translational antitermination with PTC124 (ataluren) |
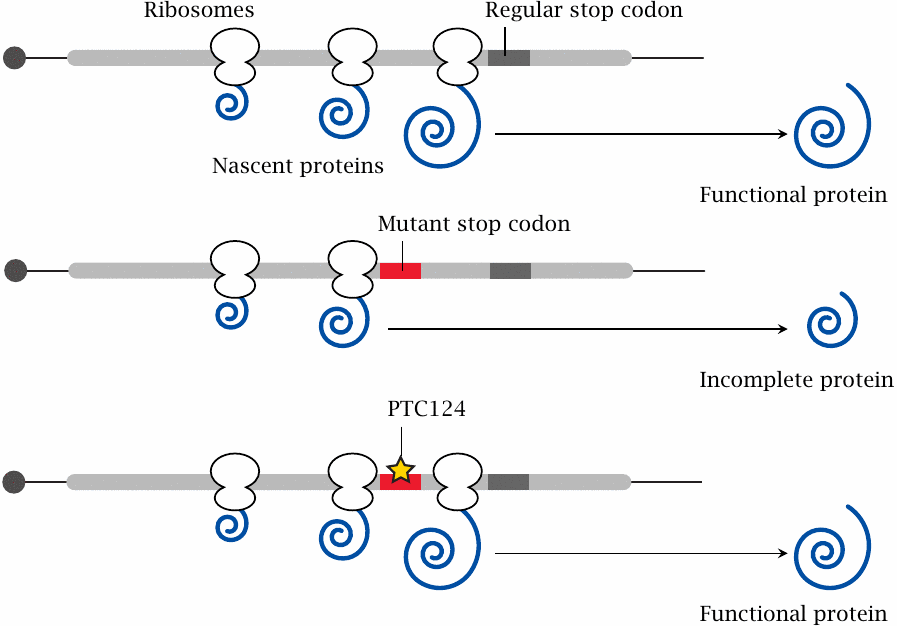
Many genetic diseases are caused by point mutations that inactivate some important enzyme or other protein. Very often, such inactivating mutations are heterogeneous; deletions, insertions, frameshift mutations, and point mutations may all give rise to the same clinical picture. The only group of patients that may benefit from translational antitermination are those who carry a point mutation that creates a premature stop codon in an otherwise intact open reading frame. In this situation, it may be possible to restore expression of a functional protein by promoting translational miscoding, that is, incorporation of some amino acid or other vis-a-vis the mutant stop codon, which then enables the ribosome to keep translating right through to the regular stop codon.132
An experimental drug that promotes translational suppression of mutant premature stop codons is PTC124 [205], also named ataluren. Its biochemical mode of action is not known exactly; however, it is noteworthy that several aminoglycoside antibiotics have similar effects, and some are also being investigated for similar therapeutic uses [206]. Aminoglycosides bind to ribosomes and, at sub-inhibitory concentrations, reduce translational fidelity; it may be that ataluren does the same.
| 20.1.4 |
Ataluren in cystic fibrosis |
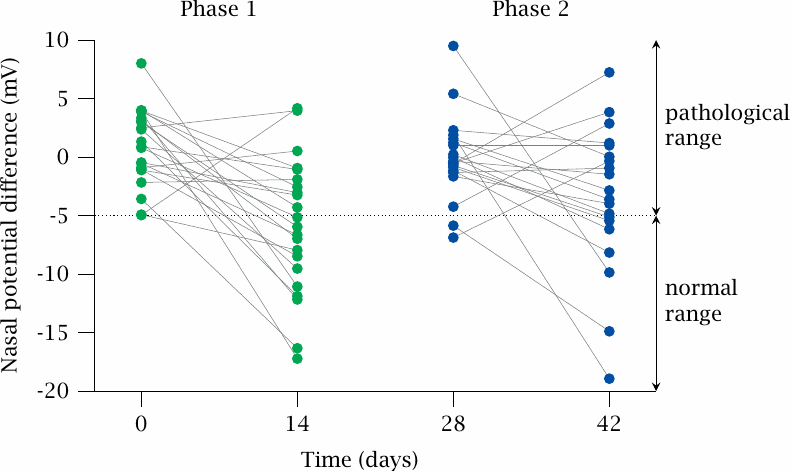
The effectiveness of ataluren in various genetic diseases has been tested in clinical pilot studies. This slide illustrates one such study, which was performed on cystic fibrosis patients. This disease is caused by a deficiency in a transporter that exports chloride ions from cells, which impedes the secretion of fluid in all kinds of exocrine glands. Multiple organs are functionally impaired, but the most serious consequences arise from the build-up of viscous mucus in the lungs, which facilitates chronic bacterial infections and ultimately leads to organ destruction.
In a sizable minority of all cystic fibrosis patients, the gene defect consists in a premature stop codon, and in this group translational antitermination is a plausible therapeutic strategy. The data illustrate the response to the drug among such patients, during two successive phases of treatment. The effect of the drug was measured as the electric potential difference that exists across the nasal mucous membrane; this potential is increased when the chloride export by the epithelial cells is reduced. Lines connect measurements on individual patients before and after each course of treatment. Evidently, most patients experienced a reduction in potential, that is, an increase in chloride export. Figure prepared from original data in [207].
| 20.1.5 |
Technical considerations for gene therapy |
- gene transfer in vivo versus in vitro
- transfer method: viral vectors vs naked DNA
- location of transferred gene: chromosomal versus episomal
- expression of transferred genes: transient versus permanent
- immune reactions to vector (particularly where repeated application is required)
The basic idea of gene therapy is to introduce a functional copy of the defective gene into the affected body cells, so that they can start making their own functional protein. In many cases, we would like to deliver the gene to only those tissues or organs that actually require the deficient gene, and to do so with high efficiency. This has in practice been difficult. However, in the example discussed below, namely, adenosine deaminase deficiency, this can be achieved by first isolating the target cells—in this case, the bone marrow stem cells—and then introducing the gene into them in vitro.
Transfer of naked DNA into cells can be achieved by electroporation or using cationic lipids. These methods are only applicable in vitro. Viral vectors, which are packaged into the viral capsids and coats proteins of some pathogenic viruses and, like regular viruses, enter body cells and deliver their nucleic acids with high efficiency, can be used both in vitro and in vivo.
One way to achieve persistence and permanent expression of the transferred gene is to stably integrate it into the genome of the recipient cell. This is most efficiently achieved with retrovirus-derived vectors, since chromosomal integration is part of the retroviral life cycle. However, a caveat is that the location of insertion within the genome cannot be reliably controlled. Integration may occur in the vicinity of some cancer-related gene, causing its transcriptional up- or downregulation. Cases of leukemia have been induced in experiments both in humans and animals.
Adenoviruses are double-stranded DNA viruses that, for the most part, replicate without integration into the host cell genome. Accordingly, adenovirus-derived vectors are less prone to insert into the genome, which makes them safer. However, such episomal replication will typically not be as long-lasting and stable as that of the retroviral vectors. If repeated application of the vector is required, this will likely work only a limited number of times, since, like the wild-type viruses, the coated vector particles are immunogenic, and antibodies will interfere with their entry into cells. Moreover, in small-scale clinical trials that used very high doses of adenovirus vectors, malignancies were again observed.
Viruses of the Herpes family remain episomal yet are persistent, which is in principle a very favorable combination. While their very large genome—Herpes viruses are second only to Pox viruses in this regard—makes their use as vectors technically challenging, they might well emerge as the vectors of choice in the long run. An overview of the state of the art concerning viral vectors can be found in [208].
| 20.2 |
Adenosine deaminase deficiency |
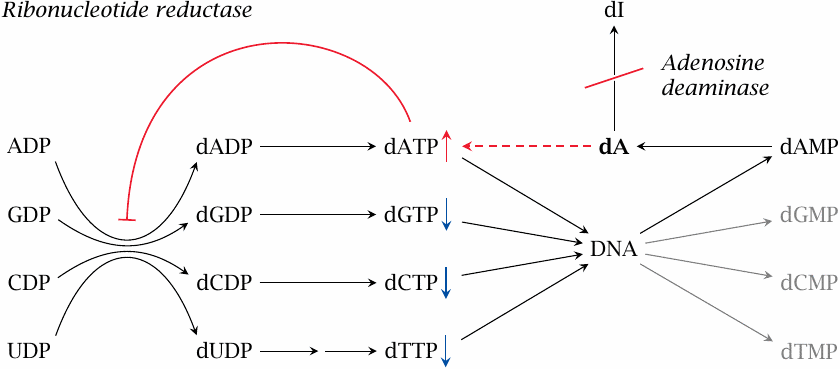
Adenosine deaminase (ADA) catalyzes the first step in the degradation of adenosine and deoxyadenosine (see section 16.5). Deoxyadenosine accumulates and is phosphorylated by salvage kinases, which yields dATP. Accumulating dATP exercises feedback inhibition on ribonucleotide reductase [209], which is required in the de novo synthesis of all deoxynucleotides (see slide 16.8).
The resulting imbalance in the supply of deoxyribonucleotides—dATP is up, whereas all others are down—constrains the rate of DNA replication.
Interference with DNA replication promotes apoptosis (see slide 19.5.1). While the enzyme defect is manifest in all cells, apoptosis actually occurs only in T-lymphocytes, which quite generally are much more prone to it than other cells. The decisive role of apoptosis in this condition is evident from the dependence of T-cell degeneration on p53 [210], a nuclear protein that is a key switch of apoptosis.
The apoptotic demise of the T-cells becomes manifest in early infancy in the form of a severe combined immunodeficiency (SCID). The deficiency is referred to as combined because it affects both humoral immunity, that is, antibody formation and cellular immunity. Antibodies are produced by plasma cells, which derive from B-lymphocytes; however, T-helper cells are required to activate the B-cells. Cellular immunity crucially depends on T-killer cells.
Without treatment, children with ADA deficiency succumb to infections within a few years; aggressive forms of treatment are therefore justified.
| 20.2.1 |
Conventional therapy of ADA deficiency: Allogenic bone marrow transplant |
- currently the standard treatment
- side effects and complications can be severe
- requires compatible donor
Lymphocytes are the only cells adversely affected by ADA deficiency. These cells originate in the bone marrow; therefore, replacement of the patient’s bone marrow with that of a healthy donor is an effective and curative treatment.
Allogenic bone marrow transplantation is a risky procedure and truly an ordeal. It requires an immunologically compatible donor. The best odds for finding one is within the family; the chance of a match between siblings is one in four. Failing that, a donor may be found anywhere in the world. As ever more people are getting typed for histocompatibility antigen profile for the sake of some transplant or other, the odds of finding a random match are improving.
However, except for identical twins,133 no donor will ever be 100% compatible, and there remains a risk of immunological complications. In addition, during the first few weeks after transplantation, the patient has essentially no immune system, and the risk of severe, even fatal infections is high. In short, less risky and less torturous alternatives are desirable.
| 20.2.2 |
ADA deficiency: an in vitro model of drug treatment |
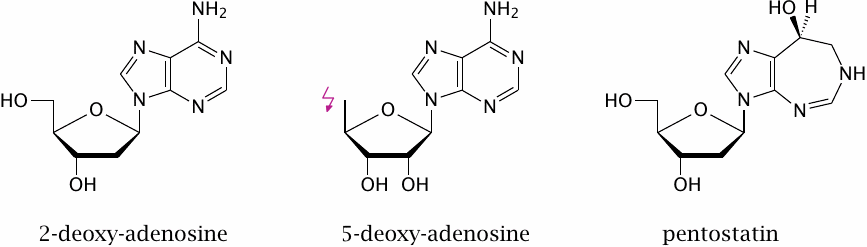
The experimental drug 5-deoxyadenosine—2′-deoxyadenosine is shown here just for comparison—inhibits deoxyadenosine kinase, which catalyzes the phosphorylation of deoxyadenosine to dAMP. Considering that the phosphorylation affects the 5-hydroxy group of the sugar, it is pretty obvious why this molecule is not a substrate of the kinase.
This inhibitor is effective in principle but not sufficient. There is a second enzyme, which is named deoxycytidine kinase but actually has a broader specificity than suggested by this name, and which also phosphorylates deoxyadenosine. In studies on cell cultures, this enzyme had to be inhibited also in order to permit lymphocytes to survive [211]. This inhibitory approach has so far only been tried in vitro; I have not yet seen any follow-up studies on its suitability in vivo.
Pentostatin is an inhibitor of adenosine deaminase. This obviously isn’t useful in treating ADA deficiency, but it can be used to create experimental animal models of the condition. It is used clinically to kill lymphocytes in graft-versus-host reactions, as well as in certain forms of lymphocyte-derived malignancies.
| 20.2.3 |
Researching ADA enzyme therapy: first attempt |
Searching PubMed for the terms in the title of this slide led me to this gem:
Priapism is a state of sustained erection without sexual arousal and is reportedly quite painful. It seems that an increased level of extracellular adenosine, which activates adenosine receptors, can induce priapism. One possible source of extracellular adenosine is the decay of red blood cells, for example in sickle cell anemia.134
According to Wen et al. [212], application of adenosine deaminase enzyme is an effective treatment for priapism. Interesting, for sure, but it has of course nothing to do with adenosine deaminase deficiency, other than that the enzyme was readily available for this therapeutic experiment because of its common use in the treatment of ADA deficiency.
| 20.2.4 |
Researching ADA enzyme therapy: second attempt |
Another interesting find was this early study:
- strategy: application of frozen irradiated red blood cells (!)
- therapy improved immune status and helped patient survive for 17 months (while waiting for blood marrow transplant)
This study [215] was performed before the advent of recombinant methods for enzyme expression and purification. The use of red cells as carriers of the enzyme is really quite ingenious! Adenosine and deoxyadenosine can leave and enter cells through nucleoside transporters (which have been discussed before, see slides 16.4.1 and 16.9.8), and can therefore undergo degradation inside the transfused red cells.
Notwithstanding the ingenuity of this treatment, it is bound to result in iron overload in the long term (see slide 17.5.3). A better form of treatment is to use the purified adenosine deaminase enzyme instead of blood transfusions. With modern means, the recombinantly expressed human enzyme would seem to be the obvious choice. At least in those patients that still express a nonfunctional version of the enzyme—as opposed to no enzyme at all; both variants occur—this enzyme should have the lowest likelihood of inducing inactivating antibodies. However, it seems that currently a bovine enzyme preparation is still in use [216]. Presumably, ADA deficiency patients are more tolerant toward the application of a non-self protein because of their immune defect. Furthermore, the bovine enzyme is modified with PEG, both to reduce its immunogenicity and to increase its dwell time in the system.135
While the inherent immunosuppression of ADA patients will facilitate the use of purified enzyme, it will also make the patients more susceptible to graft-versus-host reactions, that is, the proliferation of and immunological aggression by transferred lymphocytes. In the study cited [215], the blood cells were irradiated before transfusion, presumably in order to destroy any remaining lymphocytes and thus prevent this complication.
| 20.2.5 |
Gene therapy of ADA deficiency |
Still at the stage of clinical studies, not mainstream. A recent study [218] was performed as follows:
- Non-myeloablative conditioning
- CD34+ bone marrow cells (stem cells) were isolated from the blood, transduced in vitro with a retroviral vector carrying a functional ADA gene, and reintroduced into the body
- ADA expression achieved in lymphocytes: ~5% in bone marrow, ~75% in periphery
- All patients survived at time point of compilation of study (2–8 years after treatment), but some required additional enzyme treatment
The non-myeloablative conditioning used in this study means that the patients’ bone marrow was not entirely destroyed after the stem cells had been harvested. Therefore, after treatment, the genetically modified cells co-existed with non-modified ones.136 It is noteworthy that a full 75% of mature T-lymphocytes in the peripheral blood expressed ADA, while only 5% of the precursor cells in the bone marrow did. This discrepancy is most likely due to the survival advantage of the ADA-expressing T-cells.
My take on this cautious approach is that the investigators probably wanted to keep the option of an allogenic bone marrow transplant, in case the gene therapy failed. Myeloablative conditioning is very aggressive, and it can be used only once on each patient. Considering the fairly favorable outcome of this cautious initial experiment, it seems to me that a somewhat more aggressive approach, with a larger number of stem cells being transformed, and with a more intense decimation of the remaining bone marrow in the conditioning phase, is justified.
| 20.3 |
Enzyme therapy of lysosomal enzyme defects |
Adenosine deaminase is active in the cytoplasm, and since the substrates can enter and leave the cells by facilitated diffusion, there is no major problem with arranging for the enzyme to meet its substrate. However, this is different in lysosomal enzyme defects.
Lysosomes contain a diverse range of hydrolytic enzymes (hydrolases), which are important in the breakdown of both extracellular and intracellular molecules. These enzymes have an acidic pH optimum, which matches the acidic environment inside phagolysosomes, that is, the vesicles that form by fusion of substrate-containing vesicles with lysosomes.
There are quite a few hereditary enzyme defects that concern one or the other lysosomal hydrolase, and depending on the type of compound that now can no longer be degraded and therefore accumulates, these diseases are referred to as lipidoses, mucopolysaccharidoses, or glycogenoses.
When a lysosomal enzyme is deficient, the substrate nevertheless continues to be targeted to phagolysosomes. Therefore, in order to treat this condition with enzyme therapy, we need to get the enzyme into the lysosomes as well. We will look at some examples to understand how this can be achieved.
| 20.3.1 |
Pompe disease |
- defect of acid maltase, a lysosomal enzyme that breaks down glycogen particles
- lysosomal glycogen accumulates
- various forms: complete absence of enzyme (manifestation in infants) vs. residual activity (manifestation in older children or adolescents)
- affects mainly the skeletal muscle; glycogen accumulation leads to muscle tissue degeneration
- muscle strength progressively degrades, to the point that patients are no longer able to breathe
In Pompe disease, the deficient enzyme is acid maltase (see slide 8.6.2). Without this enzyme, lysosomal glycogen degradation stalls (see slide 8.3.7), and undegraded glycogen particles slowly accumulate. The accumulation of glycogen concerns mostly skeletal and heart muscle and leads to the degeneration of these tissues.
| 20.3.2 |
Enzyme therapy of Pompe disease |
- recombinant enzyme expressed in rabbit mammary glands, isolated from rabbit milk
- target group: juvenile patients (not infants)
- dosage: 20 mg/kg every two weeks
- clinical outcome: improvement of muscle strength, but not to normal level
- no severe immune reactions
The dosage of enzyme applied in this clinical study [219] is quite large; it is remarkable that enough of it can be obtained from rabbits. Even with this large amount of enzyme, the success of the treatment is only partial; this is most likely related to the limited efficiency of targeting the enzyme to the lysosomes.
| 20.3.3 |
Clinical outcome of enzyme therapy: Muscle strength |
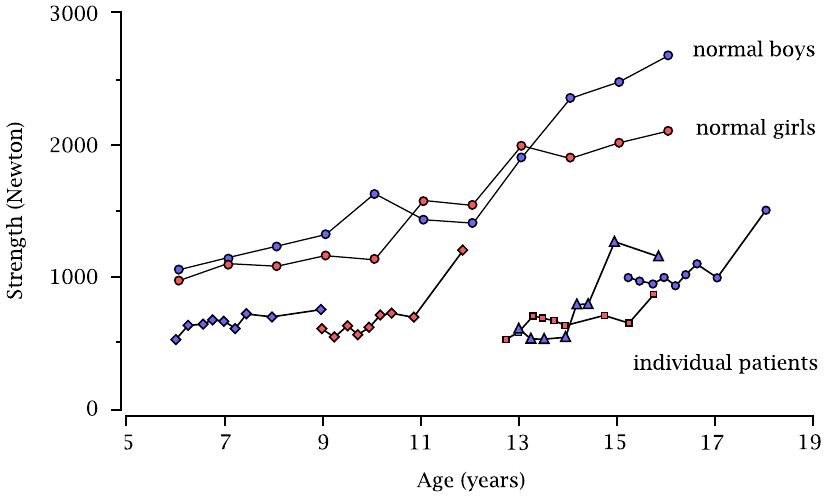
The two continuous curves at the top are the references for boys and girls. The shorter graphs at the bottom show the data from individual patients over the time course of the study. It is evident that muscle strength improves but remains below normal.
| 20.3.4 |
The mannose-6-phosphate receptor targets proteins to the lysosome |
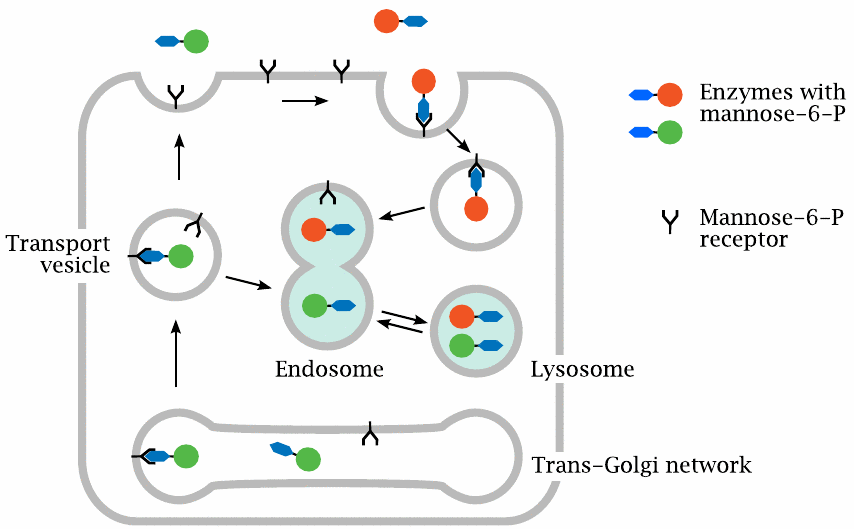
This cartoon—loosely based on one from [220]—illustrates the role of the mannose-6-phosphate receptor in targeting proteins to the lysosomes and endosomes. Proteins that contain the sugar mannose-6-phosphate in their glycosyl moieties can bind to the mannose-6-phosphate receptor, a membrane protein that occurs in several types of intracellular vesicles and compartments. The receptor picks up lysosomal enzymes within the trans-Golgi network and ensures that they indeed make their way to the lysosomes. Acid maltase is one of the proteins that are transported to the lysosomes by this mechanism.
Some mannose-6-phosphate receptor molecules also make their way to the cell surface. These receptor molecules can bind to acid maltase that was applied therapeutically and is present in the extracellular space. The receptor-enzyme complexes will undergo endocytosis and find themselves in endosomes that reversibly fuse with lysosomes. Hitching a ride inside a vagrant lysosome, an enzyme molecule may eventually alight upon the undegraded substrate within another endosome. Considering how roundabout and haphazard this transport process is, it is perhaps not surprising that the targeting of therapeutically applied lysosomal enzymes is not very efficient.138
| 20.3.5 |
Optimization of acid maltase glycosylation |
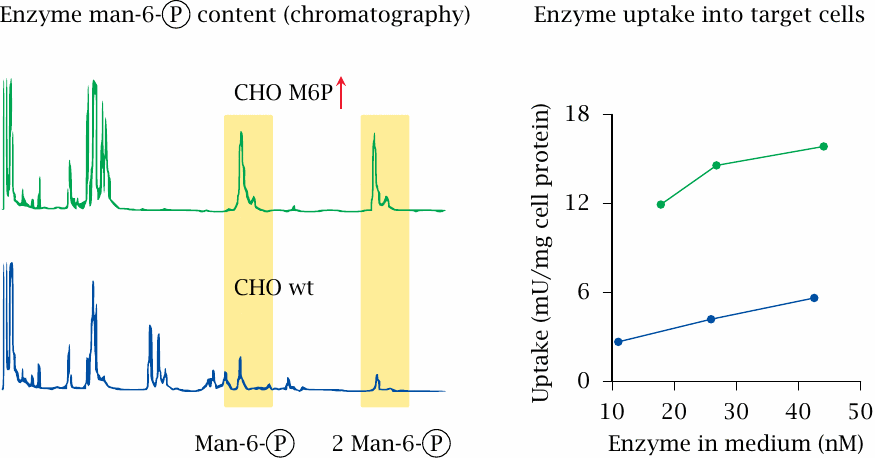
Protein glycosylation occurs within the Golgi apparatus and the trans-Golgi network. It is a stepwise process, in which several different glycosyltransferases attach sugar moieties to the ends of growing oligosaccharide chains. The composition of the oligosaccharide moieties attached to a given protein can vary with the host cell that expresses it. In the case of lysosomal enzymes, such differences may affect the rate of uptake into cells.
The experimental study summarized in this slide illustrates the importance of the carbohydrate moiety for cellular uptake. Acid maltase was expressed in control CHO cells (blue lines) and in a derived cell line engineered for increased incorporation of mannose-6-phosphate into the oligosaccharides (green lines, CHO M6P↑). The enzyme expressed in these cells contains greater amounts of mannose-6-phosphate than a control sample obtained from CHO cells (left), and it is indeed taken up more efficiently into cells lacking the enzyme (right). Figure prepared from original data in [221].
In the following slides, we will, as a final example, take a look at Gaucher disease. This is another lysosomal enzyme defect that displays some interesting and instructive differences to Pompe disease.
| 20.3.6 |
The biochemical defect in Gaucher disease |
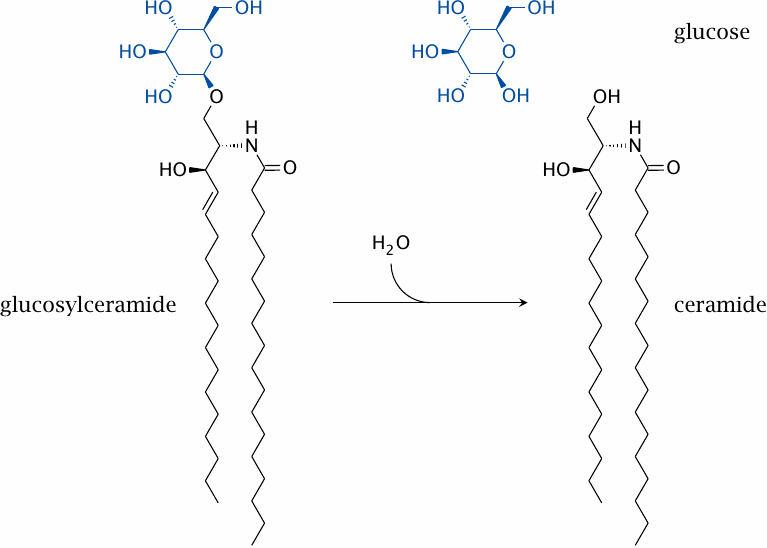
Glucocerebrosidase hydrolyzes glucosylceramide, a glycosphingolipid that occurs in cell membranes. The enzyme defect affects mostly macrophages, since these cells take up expired cells and degrade their constituents. Via an unknown mechanism, the accumulation of glucosylceramide inside the macrophages causes inflammatory, proliferative and degenerative changes in those organs that host a lot of macrophages, that is, liver, spleen, and bone marrow.
Enzyme therapy again requires the uptake of the enzyme molecules into the cells and the lysosomes. However, this is easier to achieve with macrophages than with most other cell types. Macrophages have multiple cell surface receptors that promote uptake, such as for example immunoglobulin receptors.139 Another receptor class are lectins, that is, sugar-binding proteins. Their normal function is to bind to the cell surface oligosaccharides of microbes and mediate their phagocytosis. One of these lectins, the mannose receptor, can be targeted to facilitate the uptake of therapeutically applied glucocerebrosidase.
| 20.3.7 |
Partial deglycosylation of glucocerebrosidase accelerates uptake into macrophages |
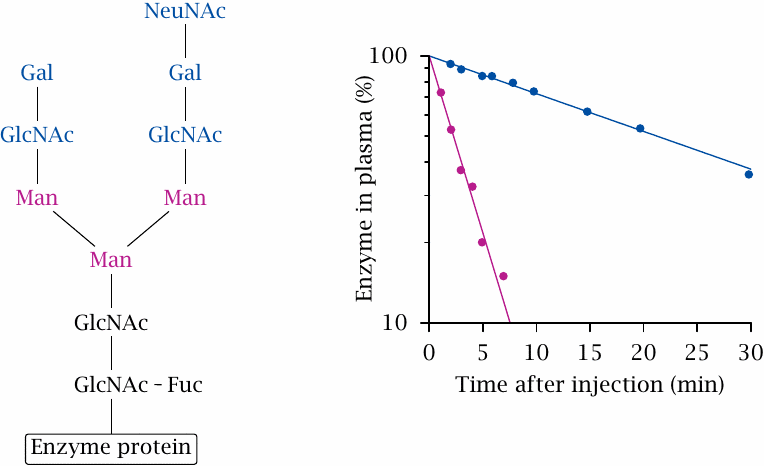
Glucocerebrosidase contains a posttranslationally attached oligosaccharide moiety that has a characteristic sequence of specific sugars. The terminal sugars—N-acetylneuraminic acid, galactose, and N-acetylglucosamine—can be removed using specific glycosidases; this will expose the mannose residues contained within and thus enable the binding of the enzyme to mannose receptors. The experiment depicted here shows that this partial deglycosylation greatly accelerates the clearance of the enzyme from plasma, such that the plasma half-life drops from 21 minutes to 2.3 minutes. Much (probably most) of the rapidly cleared enzyme winds up inside the macrophages, where it can digest accumulated glucocerebroside. Figure prepared from original data in [222].
Enzyme therapy of Gaucher disease is remarkably effective and is the current standard of treatment.
| 20.3.8 |
Drug treatment of Gaucher disease |
These two drugs are inhibitors of glucocerebroside synthesis. Total inhibition of glucocerebroside synthesis is not compatible with life, so this strategy does not obviate the need for enzyme therapy and is not free of side effects.
Serendipitously, miglustat was also found to inhibit spermatogenesis in mice, suggesting a new approach for male contraception. This effect, however, could not be substantiated in men [223].