| 19 |
Metabolism of drugs and xenobiotics |
| 19.1 |
Functional significance |
- inactivation and facilitated elimination of drugs and xenobiotics
- activation of prodrugs
- formation of active metabolites with similar or novel activity
- detoxification of toxic xenobiotics
- toxification of non-toxic xenobiotics
Drugs can be considered a subset of xenobiotics, that is, natural compounds of exogenous origin that may find their way into the human body. Other important classes of xenobiotics are potentially toxic plant alkaloids or fungal toxins. The metabolic pathways that have evolved to deal with these natural xenobiotics are active on many synthetic drugs also.
In most cases, metabolic transformation of a drug results in its inactivation and accelerated elimination from the body. However, other outcomes are possible, as will be discussed below.
| 19.1.1 |
Enzyme specificity in drug metabolism |
- key problem: a limited number of enzymes must cope with an unlimited number of substrates
- many drug-metabolizing enzymes have fairly broad specificities
- enzyme specificities overlap—many drugs give rise to multiple metabolites
The human body contains several dozen enzymes that are primarily dedicated to the metabolism of xenobiotics. Many of these enzymes can modify a large number of structurally diverse drugs and xenobiotics. In addition, a number of enzymes whose primary substrates are regular endogenous metabolites (‘eobiotics’) also participate in drug metabolism.
| 19.1.2 |
Example: metabolism of phenobarbital |
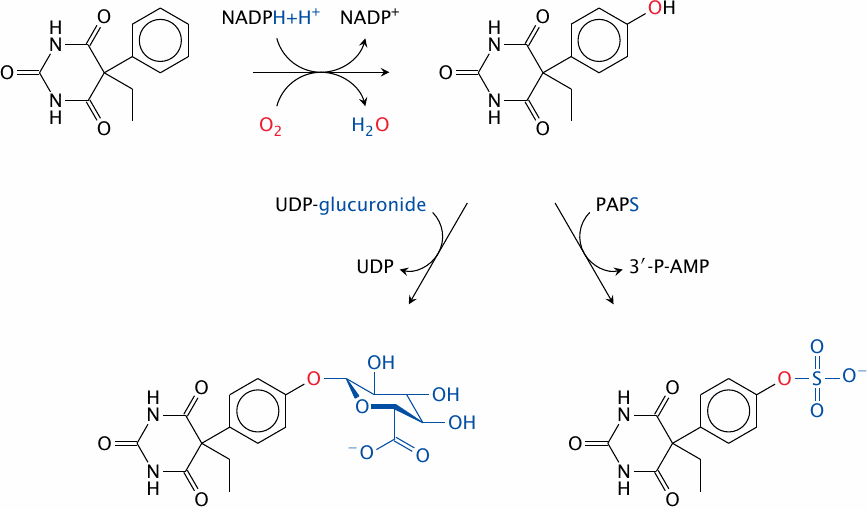
Phenobarbital, a barbituric acid derivative with both sleep-inducing and anti-epileptic activity, illustrates both the significance and the workings of drug metabolism. The drug molecule itself is quite hydrophobic. This causes the drug to distribute into fat tissue. The amount that remains in the plasma is partially bound to plasma proteins. Therefore, only a small fraction of the drug is, at any time, found freely dissolved in the blood plasma and thus amenable to filtration and excretion in the kidneys, or to biliary secretion in the liver. Elimination of the unmodified drug is thus very slow, and the lion’s share of the drug is excreted only after enzymatic conjugation.
Phenobarbital as such is not amenable to conjugation reactions. This problem is overcome by a cytochrome P450 enzyme, which introduces a hydroxyl group into the molecule. This hydroxyl group is then conjugated with either glucuronic acid or sulfate. Both of these metabolites are quite polar and are effectively excreted through the kidneys.
| 19.1.3 |
Drug metabolism facilitates drug elimination |
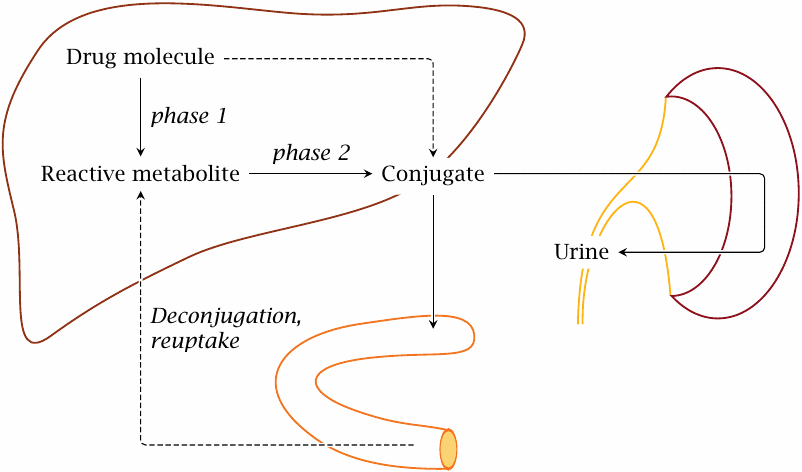
Drug metabolism works hand in hand with excretion. The key organs in drug metabolism and excretion are the small intestine, the liver, and the kidneys.
Drug-modifying enzymes are highly expressed in both the small intestine and the liver. The reactions catalyzed by these enzymes are classified into phase I and phase II reactions. Broadly speaking, a phase I reaction introduces a functional group into a substrate that enables its subsequent conjugation in a phase II reaction. Conjugation, in most cases, increases the polarity of the substrate, rendering it more amenable to secretion into the bile or to excretion in the urine. The two stages of phenobarbital metabolism depicted in the preceding slide exemplify phase I and phase II transformations, respectively.
Biliary secretion of drugs or drug conjugates requires active transport, as does tubular secretion into the nascent urine. Some of the transport proteins involved recognize the groups that were conjugated to the drug molecules, for example, glucuronic acid or glutathione. Excretion by active transport is sometimes referred to as phase III of drug elimination.
Note also that drugs that are taken orally will encounter drug-metabolizing enzymes in the small intestine and the liver before they even enter the systemic circulation. In many cases, a large fraction of the drug molecules is already modified and inactivated at this early stage; this fraction is referred to as the drug’s first pass effect.
| 19.2 |
Cytochrome P450 enzymes |
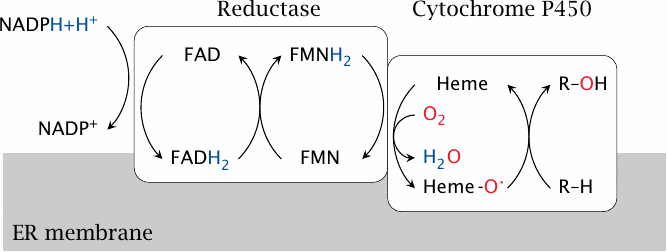
Cytochrome P450 is a large family of enzymes that are found in both prokaryotic and eukaryotic organisms. In mammalian cells, they are found in both the ER and the mitochondria, and they function in various biosynthetic pathways, such as cholesterol and steroid hormone synthesis. Several dozen CYP450 (CYP) isoforms are involved in drug metabolism; they are the most important group of enzymes in phase I metabolism. These isoforms are found mostly in the ER.
The reactions catalyzed by CYP enzymes start with the abstraction of hydrogen from NADPH by cytochrome P450 reductase, an auxiliary enzyme. The hydrogen is used by CYP to reduce one of the two atoms of molecular oxygen to water. The other oxygen atom is retained in a highly reactive form, which is then used to force one or the other kind of reaction on a substrate.
| 19.2.1 |
Reactions catalyzed by cytochrome P450 |
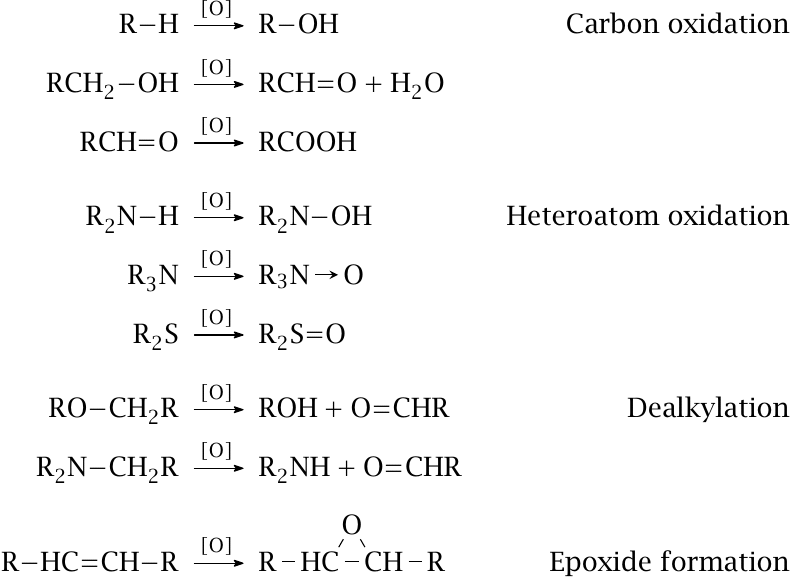
Apart from aromatic or aliphatic hydroxylation, CYP enzymes can also bring about several other types of reactions. Oxidation can occur once or repeatedly; for example, hydroxyl groups can be further oxidized to aldehydes and carboxylic acids. Oxidation is not confined to carbons but can also affect heteroatoms. Dealkylation is a powerful way to break up and inactivate drug molecules.
Amine oxidation, aldehyde formation, and epoxide formation yield reactive groups that may subsequently cause toxic effects. Therefore, while drug metabolism often abolishes toxicity, sometimes is can actually create it.
| 19.2.2 |
Transcriptional induction of CYP450 3A4 |
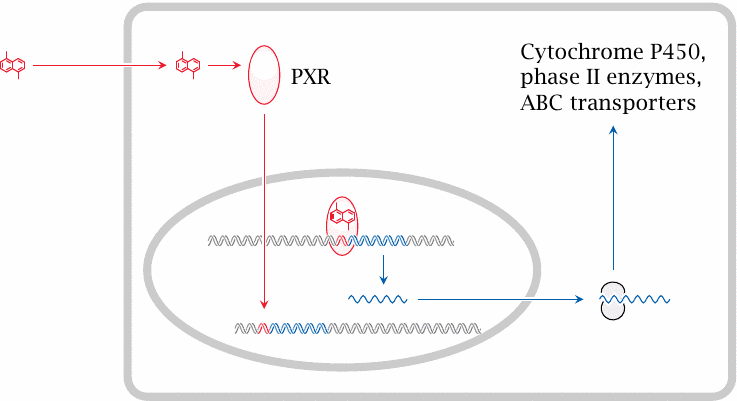
Considering that there are several dozen different CYP enzymes in human cells, it is remarkable that a single isoform, CYP3A4, is involved in the metabolism of some 50% of all clinically prescribed drugs. Like several other isoforms, CYP3A4 is inducible, that is, its rate of gene transcription is increased by certain drugs.
With CYP3A4, the nuclear hormone receptor that mediates induction is the pregnane X receptor (PXR). When a suitable drug binds to this receptor, it translocates from the cytosol to the nucleus and binds to its cognate regulatory DNA sequences, referred to as xenobiotic response elements (XRE); this results in increased transcription of genes in the vicinity. The PXR responds to a particularly wide variety of drugs, which accounts in part for the prominent role of CYP3A4 in drug metabolism.
Enzyme induction often leads to accelerated metabolism of multiple drugs, not just the inducing drug itself. Examples are rifampicin, an antibiotic used in tuberculosis, as well as phenytoin and phenobarbital, which are used as anti-epileptic agents. These drugs all induce accelerated inactivation of each other. Furthermore, they also accelerate the metabolism of contraceptives and render these drugs ineffective.
Together with cytochrome P450 enzymes, conjugating enzymes and active transporters that function in drug excretion are also induced. CYP enzymes contain heme; the first and rate-limiting step of heme synthesis is catalyzed by δ-aminolevulinate synthase (see slide 17.2.2). This enzyme is also induced, and through this mechanism certain drugs, including the examples listed above, can exacerbate acute intermittent porphyria (see slide 17.3.6).
| 19.2.3 |
Structure of erythromycin bound to cytochrome P450 3A4 |
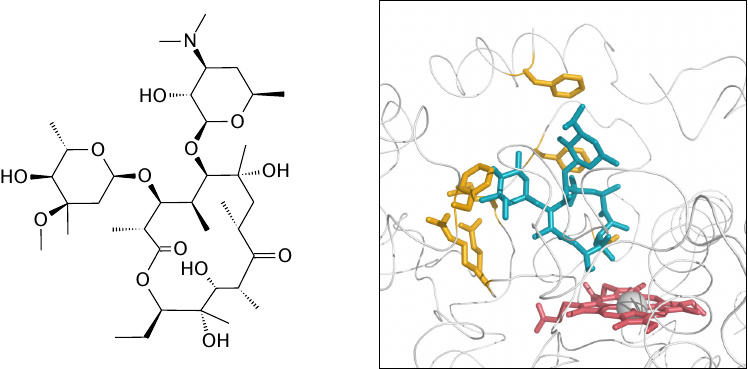
Another reason of CYP3A4’s prominent role in drug metabolism is its own ability to accommodate a wide variety of substrates within its active site. This slide and the next one illustrate two examples. The drugs, erythromycin in this slide and ketoconazole in the next one, are shown in blue or green. The heme of CYP3A4 is shown in red, and several amino acid side chains that interact with the drug molecules are shown in yellow. Note the differences in protein conformation and the interacting residues between both slides.
| 19.2.4 |
Ketoconazole bound to cytochrome P450 3A4 |
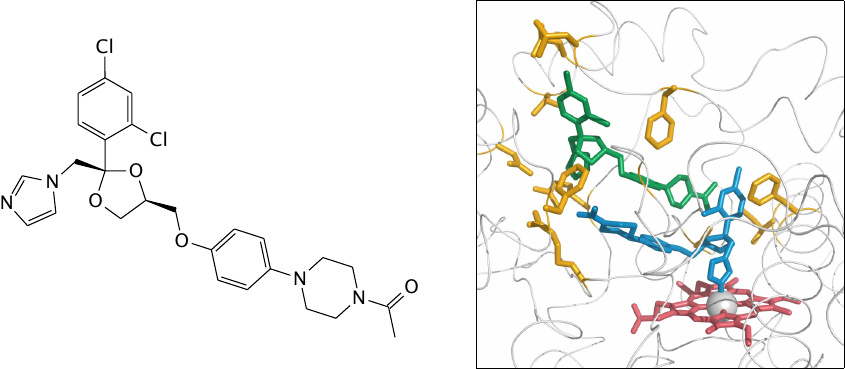
This slide shows two molecules of ketoconazole bound within the active site of CYP3A4. The binding of the second molecule is likely an artifact of the high drug concentration used in the crystallography experiment.
Note that the imidazole group of one drug molecule is bound to the heme iron. Ketoconazole is an antifungal agent that inhibits 14α-demethylase, a cytochrome P450 enzyme that is essential for the synthesis of ergosterol, the major sterol found in fungal cell membranes. From this mode of action, we can understand that, as a side effect, ketoconazole also inhibits drug metabolism in humans.
| 19.2.5 |
Superposition of the erythromycin- and the ketoconazole-bound structures |
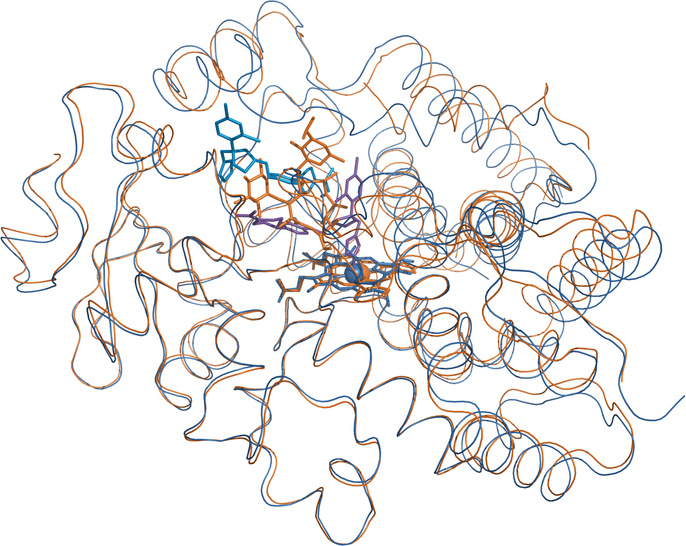
The erythromycin-bound structure is shown in orange, and the ketoconazole-bound one in blue. The polypeptide backbones track each other closely for the most part, but they diverge noticeably in several places, particularly atop the active site. These local deviations illustrate the remarkable conformational flexibility of the enzyme molecule that allows it to accommodate, and therefore to metabolize, a large number different substrates.131
| 19.2.6 |
Examples of active metabolites formed by CYP450 enzymes |
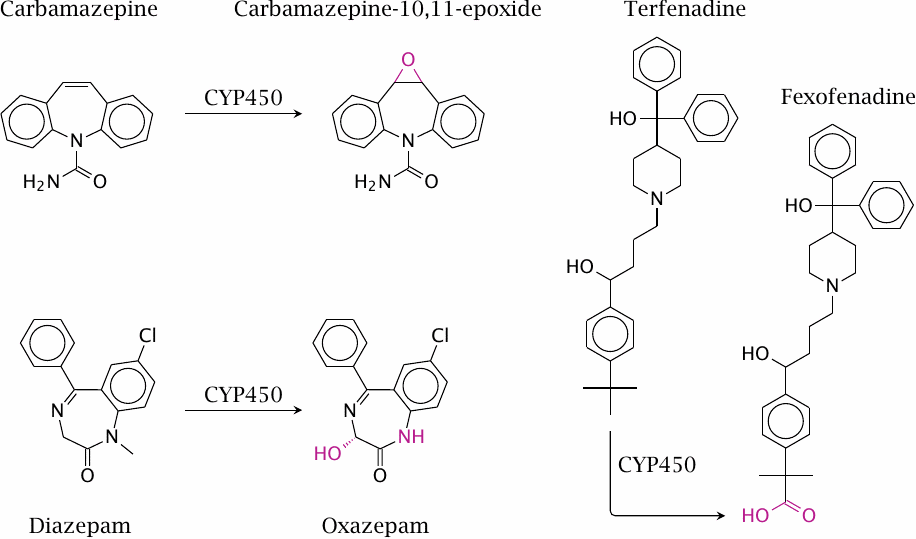
While drug metabolism often results in inactivation, metabolites may retain pharmacological activity, or sometimes even acquire novel ones. For example, two CYP-mediated reactions convert diazepam to oxazepam, which retains the pharmacological activity of the parent compound.
Some active metabolites, including oxazepam and fexofenadine, have become drugs in their own right. Since these molecules are already prepared for conjugation, they are usually more rapidly eliminated than the parent compounds. In the case of oxazepam, this is an advantage when the intention is to induce sleep, since most of the drug will have been excreted the next day. In contrast, diazepam works better in the treatment of epilepsy, since in this application a more stable and steady level of drug activity is desired.
With all examples shown in this slide, both the parent molecules and the metabolites have pharmacological activity. For various applications, drugs have been designed that actually require metabolic conversion to become active; such molecules are referred to as prodrugs. Some organic nitrates, for example nitroglycerin, are metabolized by cytochrome P450 enzymes and by mitochondrial aldehyde dehydrogenase to release nitric oxide as the active principle. Other examples of prodrugs that we have already encountered are sulfamidochrysoidine (see slide 15.3) as well as tenofovir and cidofovir (slide 16.9.14).
| 19.2.7 |
Benzopyrene as an example of harmful metabolism of xenobiotics |
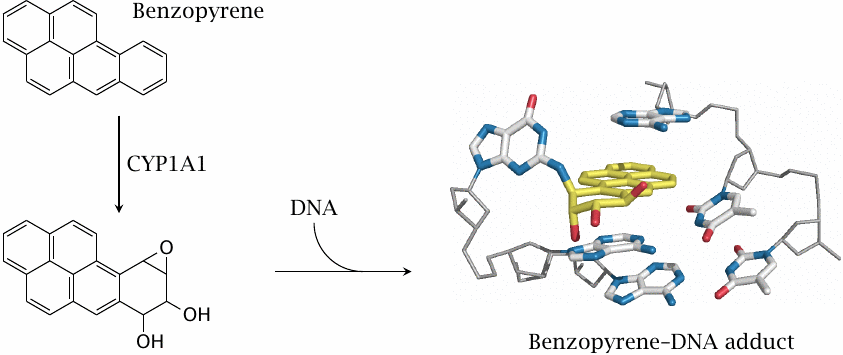
Benzopyrene and related compounds activate the aromatic hydrocarbon receptor (AHR), a nuclear hormone receptor that is functionally similar to PXR but instead of CYP3A4 induces CYP1A1. The reaction of this enzyme with aromatic hydrocarbons tends to produce epoxides, which react easily with nucleophiles. Epoxides of polycyclic aromatic molecules are particularly harmful, since they can intercalate into DNA. The epoxide can then react covalently with the DNA and cause mutagenic damage.
Polycyclic aromatic hydrocarbons such as benzopyrene arise from incomplete combustion of organic matter, such as in car exhaust fumes or cigarette smoke. Their metabolic conversion to epoxides and subsequent reaction with DNA is the major mechanism of carcinogenesis in smokers.
| 19.3 |
Phase II reactions |
| 19.3.1 |
Summary of phase II reactions |
| Enzymes | Cosubstrates | Functional groups |
| UDP-glucuronosyltransferases | UDP-glucuronide | –OH, –NH2 |
| sulfotransferases | PAPS | –OH, –NH2 |
| glutathione-S-transferases | glutathione | epoxy groups, double bonds |
| acetyltransferases | acetyl-CoA | –OH, –NH2 |
| methyltransferases | SAM | –OH, –NH2, –SH |
| epoxide hydrolase | H2O | epoxide groups |
| aminoacyltransferases | amino acids | –COOH |
This list is sorted roughly according to decreasing importance. Glucuronidation is the most common reaction, followed by sulfation; both were illustrated in slide 19.1.2. Some more reactions will be illustrated below.
None of the cosubstrates and reaction mechanisms employed in conjugation are used exclusively in drug metabolism. UDP-glucuronide and PAPS are also used in the synthesis of proteoglycans, glutathione occurs in many redox reactions and in the synthesis of leukotriene mediators, and amino acid conjugation is used with bile acids. Some enzymes may act upon both endogenous metabolites and xenobiotics; examples are catechol-O-methyltransferase, which apart from drugs also conjugates epinephrine and norepinephrine, and glycine-N-acyltransferase, which attaches glycine to both drugs and to cholic acid.
| 19.3.2 |
Detoxification of benzopyrene epoxide derivatives by epoxide hydrolase or glutathione-S-transferase |
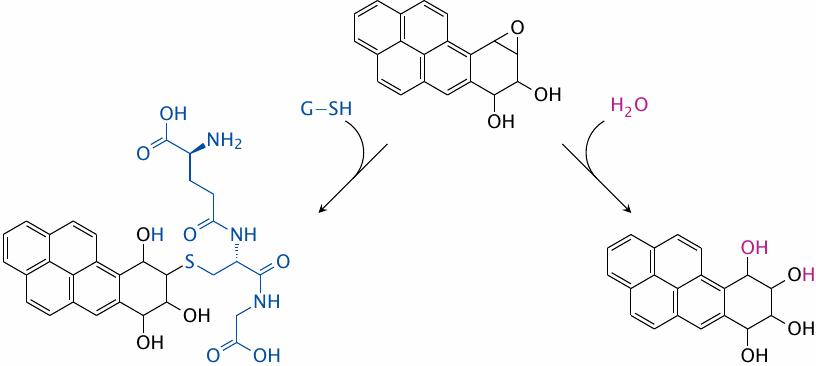
Fortunately for smokers, not all aromatic epoxide molecules will end up reacting with DNA. One major detoxification pathway is the reaction with glutathione, which is facilitated by glutathione-S-transferase (left). Epoxide hydrolase (right) also contributes to the detoxification.
The epoxide hydrolase reaction occurs after a phase I reaction, so therefore could be considered part of phase II. On the other hand, it does not result in conjugation, and it thus might be considered not to be part of phase II. So, which phase does it belong to?
This question is purely one of definition and therefore irrelevant. It is only mentioned to illustrate that the distinction between phase I and II reactions, while useful, has its limitations.
| 19.3.3 |
Metabolism of acetaminophen |
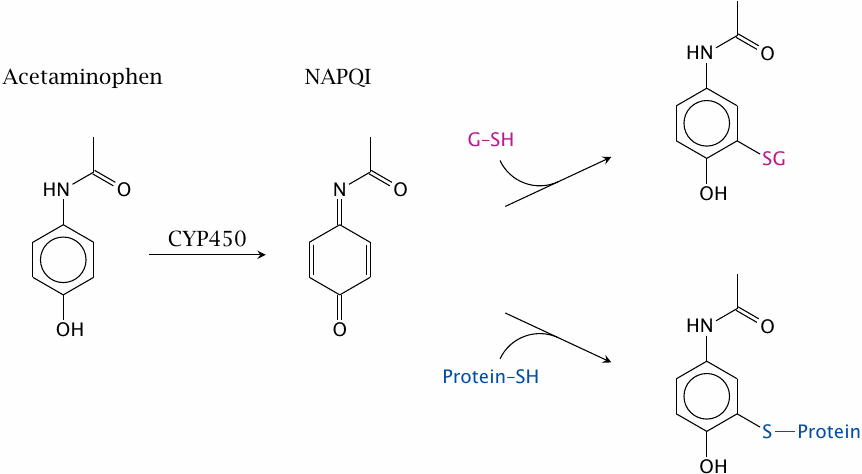
Acetaminophen also undergoes successive phase I and phase II reactions. The initial CYP-catalyzed reaction yields N-acetyl-p-benzoquinone imine (NAPQI). This molecule is also quite reactive towards nucleophiles, particularly sulfhydryl groups. Glutathione is the most abundant intracellular thiol, with a free concentration of ~5 mM, and while supplies last will neutralize most NAPQI. However, once glutathione has been depleted, NAPQI will start reacting with cellular macromolecules and cause cytotoxicity. This mostly affects the liver, since it has the highest activity of cytochrome P450 enzymes and therefore will produce the most NAPQI.
Acetaminophen is well tolerated when applied at dosages that will not deplete glutathione. However, it turns toxic rapidly once the safe dosage limit is exceeded.
| 19.3.4 |
Morphine skips phase I and is conjugated directly |
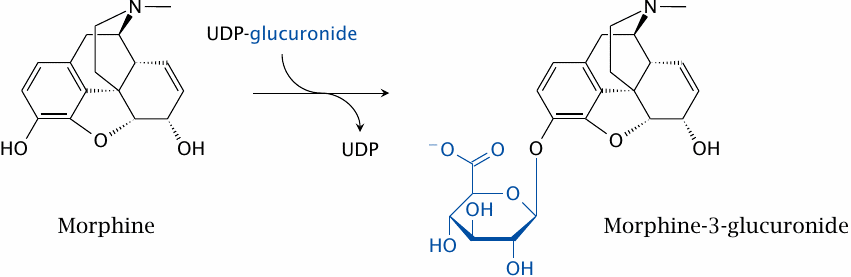
Phase I reactions are not necessary if a drug molecule already contains functional groups suitable for conjugation. An example is morphine, which has two hydroxyl groups. The conjugation of either, or both, with glucuronic acid is sufficient for excretion. Interestingly, one of the two single glucuronides—the one not shown here—retains pharmacological activity.
N-demethylation of morphine by cytochrome P450 can occur but does not significantly affect excretion.
| 19.3.5 |
Acetylation of INH by N-acetyltransferase 2 (NAT 2) |
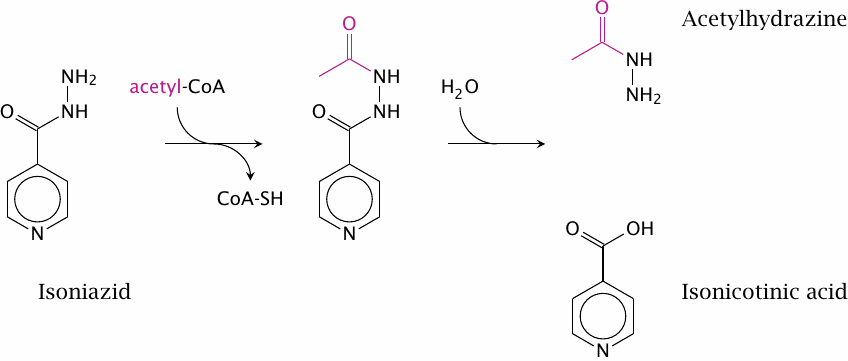
The metabolism of isoniazid (isonicotinic acid hydrazide, INH; a tuberculostatic agent) also starts with a phase II reaction. N-acetyltransferase 2 (NAT2) acetylates the hydrazide group. The product can decay, and the acetyl-hydrazide can transfer its acetyl group to other nucleophiles in the cell.
| 19.3.6 |
Bimodal distribution of INH acetylation speed |
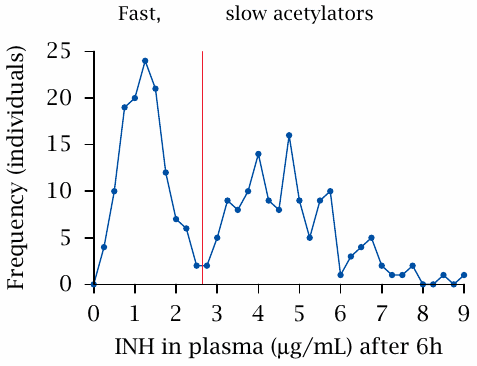
The depicted experiment measured the speed of acetylation. A fixed test dose was applied at t = 0, and the remaining plasma concentration was determined six hours later. There clearly are two separate peaks, which represent the fast and slow acetylators, respectively. Among Caucasians, about 50% express an inactive NAT2 allele, which causes the slow-acetylator phenotype. The percentage of slow acetylators is lower among Asians (but was higher in a small study on Kenyans [200]; I don’t know how representative that study is).
Apart from isoniazid, the NAT2 enzyme and its polymorphism also affects the inactivation rates of some other drugs, such as procainamide and hydralazine, which in slow acetylators are more likely to cause toxicity. A role of this polymorphism has also been reported in the susceptibility to bladder cancer caused by aromatic amines, which in Europeans was found to correlate with slow acetylator status. Surprisingly, this correlation was not observed in Chinese [201]. The reason for this discrepancy seems to be unknown.
| 19.3.7 |
Metabolic activation of arylamine carcinogens |
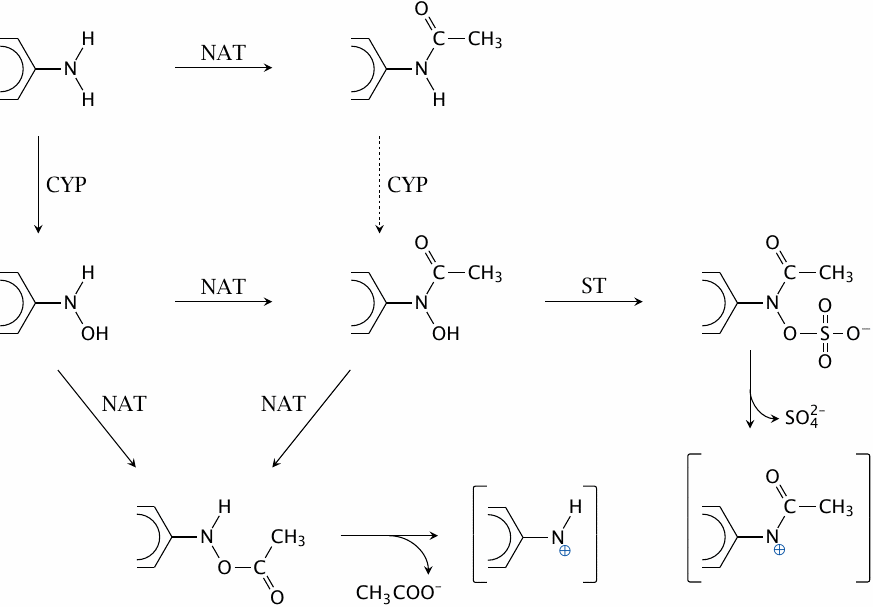
N-Acetyltransferases (NAT), cytochrome P450 (CYP) and sulfotransferases (ST) cooperate in the metabolic activation of arylamine carcinogens such as benzidine or 2-naphthylamine. The acetoxy and sulfohydroxamate products decay spontaneously to reactive electrophiles, which can then react covalently with cellular macromolecules, including DNA.
The activation is most efficient if CYP acts before NAT; acetylation of arylamines therefore affords partial protection from carcinogenic activation. This likely accounts for the lower susceptibility of fast acetylators to arylamine-induced tumors. Figure drawn after a scheme shown in [202].
| 19.3.8 |
Amino acid conjugation: Glutamine conjugation of phenylacetate |
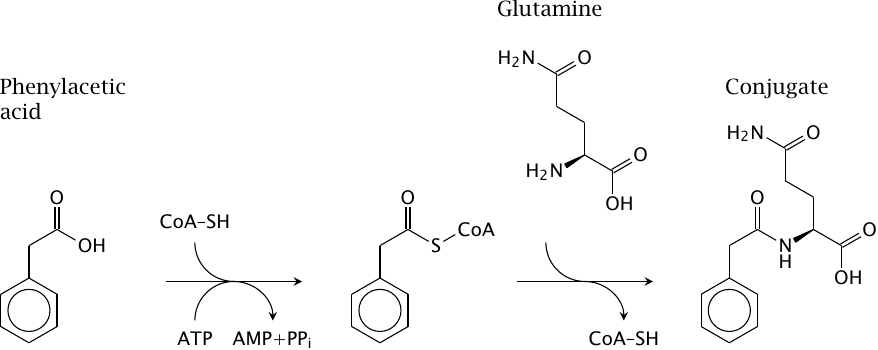
Amino acid conjugation is limited to xenobiotics that are carboxylic acids, which are first activated to coenzyme A-thioesters and then linked to an amino acid by an amide bond. The amino acid may be glutamine (as shown), glycine, or taurine. The latter two amino acids are also used in the synthesis of the conjugated bile acids taurocholate and glycocholate (see slide 11.5.1).
The pathway can be put to use in the alternate pathway therapy of urea cycle enzyme defects (see slide 12.3.10). In this application, the usual physiological role of amino acid conjugation is turned on its head. Normally, amino acids are expended in order to eliminate unwanted organic acids; in contrast, here we supply innocuous organic acids, the amino acid conjugates of which then become vehicles for the elimination of surplus nitrogen. Brilliant!
An organic acid commonly used in alternate pathway therapy is phenylbutanoic acid, which first undergoes β-oxidation to phenylacetic acid (the substrate shown here) before conjugation to glutamine and excretion. It may be combined with benzoic acid, which undergoes conjugation with glycine and therefore recruits another enzyme and another pool of nitrogen.
| 19.4 |
Reductive drug metabolism |
Multiple enzymes:
- methemoglobin reductase (diaphorase)
- cytochrome P450 reductase
- thioredoxin
- bacterial metabolism
- …
While cytochrome P450 enzymes play the most important role in phase I metabolism, some drugs are actually reduced rather than oxidized at this stage. The enzymes involved are a somewhat heterogeneous bunch and primarily serve in roles other than the metabolism of xenobiotics.
| 19.5 |
Anti-tumor drugs that are preferentially activated in tumor cells |
This section, after some introduction, examines two antitumor prodrugs that attempt to exploit metabolic activation to selectively target tumor cells.
| 19.5.1 |
DNA damage triggers programmed cell death |
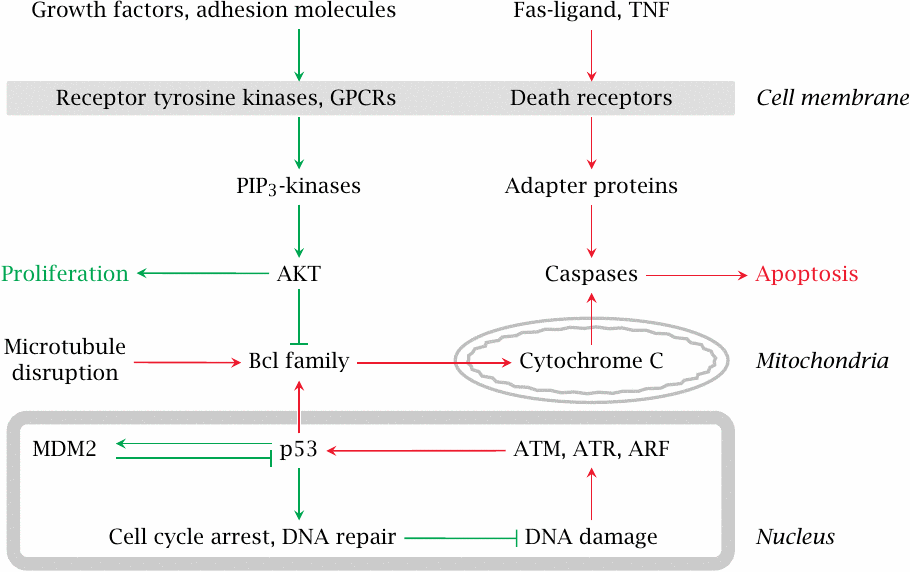
Programmed cell death, or apoptosis, is a protective mechanism that inhibits the proliferation of genetically damaged cells. DNA damage is detected by so-called checkpoint proteins during various stages of the cell cycle. Extracellular signals can promote or inhibit apoptosis. Cancer cells are often more susceptible to apoptotic stimuli than normal cells are. This is exploited by the use of DNA-damaging, cytotoxic drugs in cancer chemotherapy.
| 19.5.2 |
Mechlorethamine, a DNA-alkylating drug |
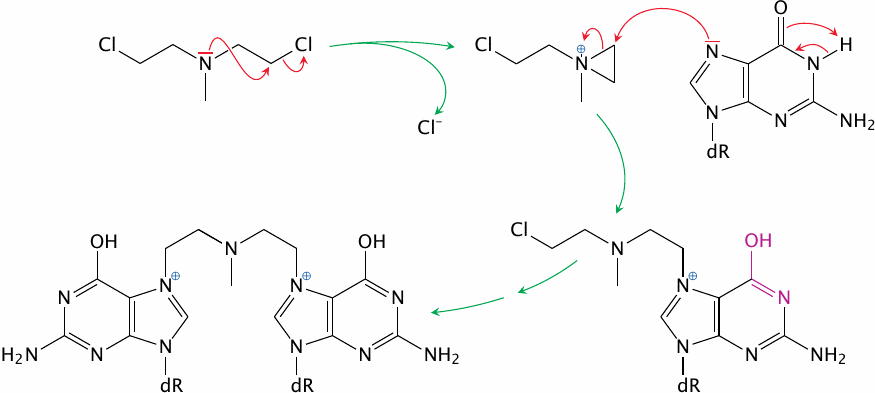
Many anticancer drugs are DNA-alkylating agents; mechlorethamine is a straightforward example. Bifunctional DNA-alkylating agents such as this one have much higher antitumor activity than monofunctional ones, since they can crosslink the two DNA strands; this makes semi-conservative DNA repair impossible.
| 19.5.3 |
CB 1954, an experimental antitumor drug that is activated by nitro group reduction and acetylation |
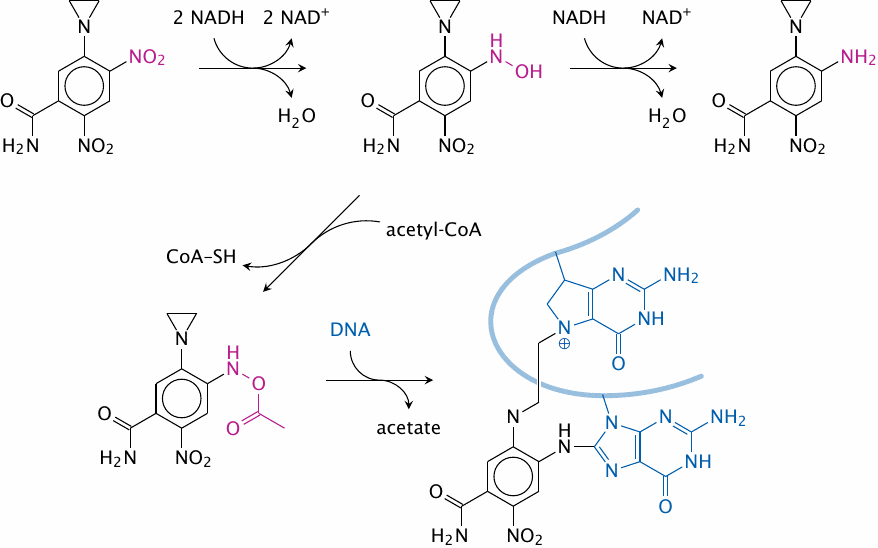
The experimental drug CB 1954 is, in its original form, a monovalent alkylating agent; the alkylating group is the aziridine group (the three-membered ring at the top). It is activated to a bifunctional agent in two steps. Diaphorase (methemoglobin reductase) reduces the nitro group to a hydroxylamine, which is then acetylated by an acetyltransferase to form a reactive acetoxy group [203].
For reasons that are not well understood, the environment inside tumor cells is often more reducing than in nontumorous cells, which will result in preferential activation of this drug inside tumor cells. CB 1954 is somewhat of a perennial “experimental” drug, which probably means that it is not very compelling in clinical use. However, several established anticancer drugs such as doxorubicin and bleomycin also require reductive activation [204], which may enhance their selectivity for cancer cells.
| 19.5.4 |
Canfosfamide, an antitumor drug that targets alkylant-resistant tumor cells |
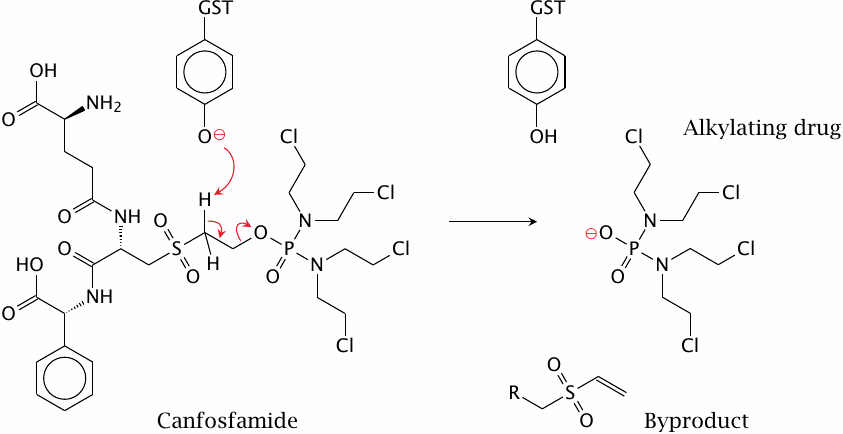
Glutathione conjugation can inactivate alkylating agents, for example ones with epoxide groups (slide 19.3.2) or anticancer drugs such as mechlorethamine (slide 19.5.2). Accordingly, tumor cells may develop resistance to alkylating antitumor drugs by increasing their expression of glutathione-S-transferase.
The antitumor agent canfosfamide targets such cells, since it has been designed to require activation by glutathione-S-transferase (GST; the catalytic residue is a tyrosine). This drug is currently in clinical trials.