| 8 |
Glycogen metabolism |
| 8.1 |
Overview |
In addition to gluconeogenesis, the reversible storage of glucose in the form of glycogen provides a second major mechanism of glucose homeostasis.
Glycogen is a branched glucose polymer that is found in many organs, but the largest quantities occur in the liver and in skeletal muscle. The liver can store up to 150–200 grams, which amounts to 10% of the organ’s wet weight. It draws from this reservoir to maintain the blood glucose concentration; glycogen plays a major role in day-to-day glucose homeostasis.
While skeletal muscle contains glycogen at much lower concentration than the liver, its much larger overall mass means that the absolute amount of glycogen stored there is approximately twice higher than in the liver. The contribution of muscle glycogen to glucose homeostasis is less well understood.
While liver and skeletal muscle store the lion’s share of glycogen, it also occurs in other organs such as the heart, the brain, and the kidneys. All of these organs may therefore be affected by glycogen storage diseases (see Section 8.6.
| 8.2 |
Glycogen structure |
| 8.2.1 |
Why store glucose in polymeric form? |
-
The osmotic pressure is governed by the gas equation:
\[ pV = nRT \: \Longleftrightarrow \: p = \frac{n}{V}RT \nonumber\] - Glycogen amounts to 10% of the liver’s wet weight, equivalent to 600 mM glucose
- When free, 600 mM glucose would triple the osmotic activity of the cytosol—liver cells would swell and burst
- Linking 2 (3, …) molecules of glucose divides the osmotic effect by 2 (3, …), permitting storage of large amounts of glucose at physiological osmolarity
The proportionality of concentration and osmotic activity does not strictly apply to large molecules, but the approximation is good enough for the present purpose.
| 8.2.2 |
Covalent structure of glycogen |
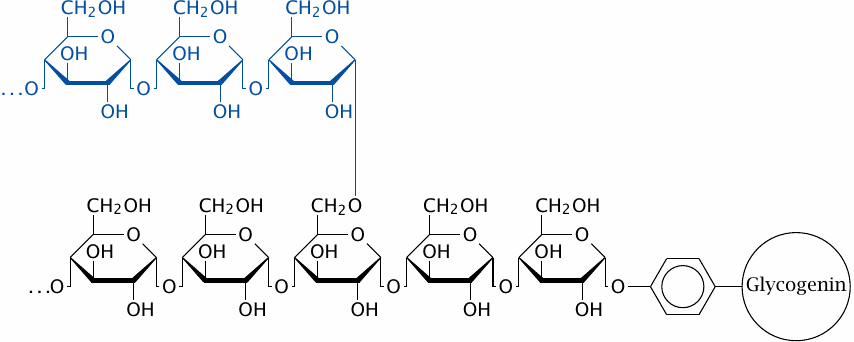
Glycogen consists of linear stretches of glucose residues connected by α-1→4-glycosidic bonds, with branches that are attached through α-1→6-glycosidic bonds. The entire tree-shaped polymer, or dendrimer, is rooted in a single molecule of the protein glycogenin.44 Each linear stretch contains approximately 13 glucose residues and, except of course for the outermost layer of the molecule, carries two branches of the same length that are attached 3–4 residues apart.
The structure of glycogen is similar to that of amylopectin (see slide 1.6.11). However, in glycogen, the density of branches is greater, which means that a glycogen molecule has a greater number of free ends than an amylopectin molecule of the same molecular weight. The number of free ends determines the possible rates of synthesis and breakdown, and the greater number of free ends in glycogen than in amylopectin reflects a difference in metabolic rates, which are higher in animals, particularly warm-blooded ones, than in plants.
| 8.2.3 |
The size of glycogen particles is limited by crowding in the outer layers |
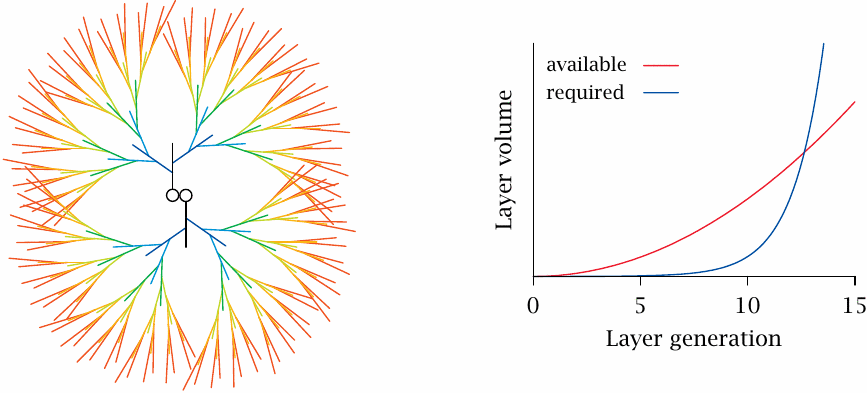
According to the rules detailed in the preceding slide, the number of branches will double with each successive generational layer of the glycogen molecule. However, the amount of space available to those branches will only grow in proportion to the square of the particle radius or, approximately, the square of the number of generations. Taking into account the actual dimensions and architecture of the polymer, it has been calculated that in the 13th generation the required space would exceed the available space. Therefore, a glycogen molecule can contain no more than 12 generational layers. This implies that a single glycogen molecule can contain up to approximately 54,000 glucose residues; it will have a molecular weight of almost 107 Da and a diameter of approximately 25 nm [32].
Electron microscopy shows glycogen particles whose dimensions agree well with this theoretical maximum size of single molecules; these are referred to as β particles. In many tissues, variable numbers of β particles are found clustered into so-called α particles. Interestingly, α particles can be broken up with thiol-reducing agents, which implies that they are held together by disulfide bonds. Disulfides usually form between protein molecules. In addition to such scaffolding proteins, glycogen particles also contain a considerable number of enzymes and regulatory proteins [33], some of which will be discussed below.
| 8.2.4 |
Glycogen is more loosely packed and more soluble than amylose |
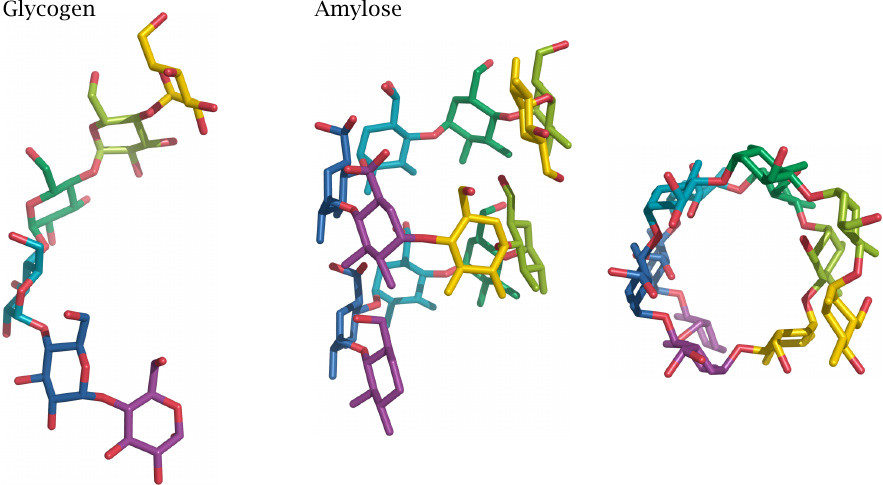
The above structural model of glycogen assumes a relatively loose packing of the glucose residues within the α-1→4-linked linear stretches. However, perfectly linear polyglucose—that is, amylose—adopts a much more densely packed helical structure. In this structure, more hydroxyl groups are engaged in hydrogen bonds with other glucose residues rather than with water; amylose therefore has low aqueous solubility.45
Unlike amylose, which mostly serves for long-term storage in plant bulbs and seeds, glycogen typically is degraded within hours of synthesis; this rapid turnover is facilitated by its loose structure. However, if branch formation breaks down, aberrant, condensed glycogen particles may arise that are no longer amenable to regular turnover. Various enzyme defects that interfere with branch formation cause the accumulation of such particles inside the cells; an example is the defect of the enzyme laforin in Lafora disease (see slide 8.6.4).
Aberrant glycogen particles also arise spontaneously in normal metabolism. The high density of polyglucose chains in the outermost layers of the glycogen molecule may interfere with the activity of branching enzyme and may promote tighter packing. Lysosomal glycogen degradation (see section 8.3.7) may have evolved as a pathway to dispose of such dysfunctional particles.46
| 8.3 |
Glycogen synthesis and degradation |
Synthesis:
- synthesis of an activated precursor, UDP-glucose, by UTP:glucose-1-phosphate uridylyltransferase
- initiation of glycogen synthesis by glycogenin
- introduction of branches by branching enzyme
- chain elongation by glycogen synthase
- repeat steps 3 and 4
Degradation:
- depolymerization of linear strands by phosphorylase
- removal of branches by debranching enzyme
- repeat steps 1 and 2
This slide summarizes the enzyme reactions that occur in glycogen synthesis and degradation, respectively. As you can see, the regular, periodic structure of glycogen corresponds to similarly regular and periodic methods of synthesis and breakdown that require only a small number of different enzymes.
| 8.3.1 |
Activation of glucose for glycogen synthesis |
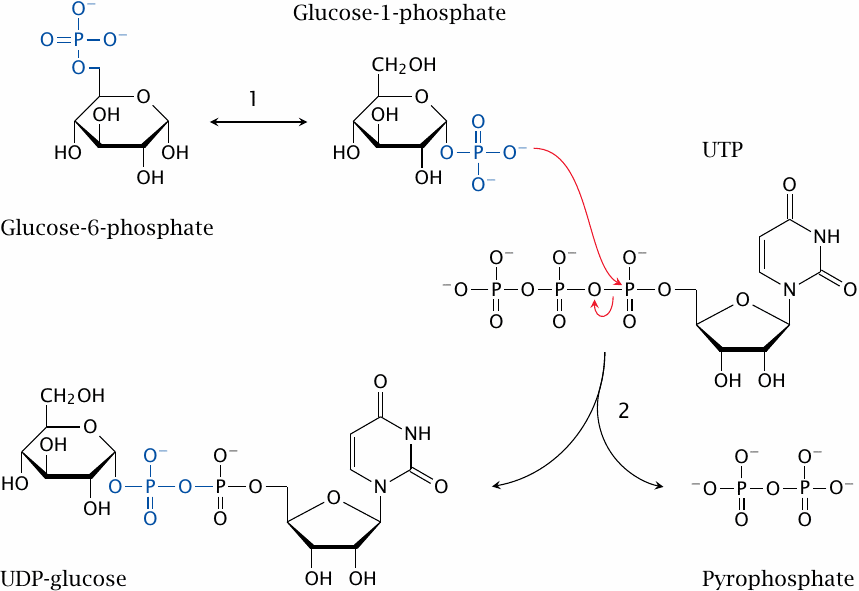
Both glycogenin and glycogen synthase use an activated form of glucose, UDP-glucose, which is formed from glucose-6-phosphate in two steps. Phosphoglucomutase first transforms glucose-6-phosphate to glucose-1-phosphate (1), which is then converted to UDP-glucose (2). The latter reaction requires uridine triphosphate (UTP) and releases pyrophosphate.47
The UDP-glucose that is used in the Leloir pathway of galactose degradation (slide 4.3.1) is derived in the same manner. UDP-glucose is also the precursor of UDP-glucuronic acid, which is used in the conjugation of bilirubin (section 17.4) and of xenobiotics (section 19.3).
| 8.3.2 |
Overview of glycogen synthesis |
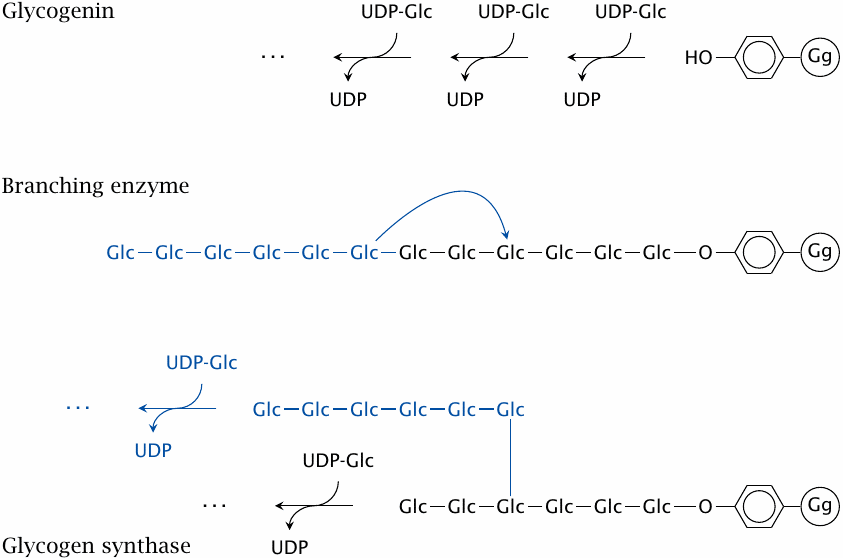
Glycogenin is a small bifunctional protein that serves both as the starter substrate and the polymerase that synthesizes the initial linear strand of glucose residues. It begins the synthesis by attaching the initial glucose residue to a strategic tyrosine (Y194) side chain of itself, and then successively adds several more glucose residues to the sugar’s 4′ end.
The linear chain synthesized by glycogenin may contain up to ~12 glucose residues. This chain length would be long enough to serve as a substrate for branching enzyme, and it seems likely that the next step is indeed the introduction of the first branch. Alternatively, it is possible that the free end is first extended some more by glycogen synthase before branching occurs. In any event, after branching has occurred, glycogen synthase extends both of the two available 4′ ends. The remainder of the molecule is built through the alternating actions of branching enzyme and glycogen synthase; the cycle repeats until the glycogen reaches its inherent size limit at approximately 10 MDa (see slide 8.2.3).
The reactions performed by glycogenin itself and by glycogen synthase are equivalent. It is interesting to note that the carbon 1 of each glucose subunit is in the α-configuration both in UDP-glucose and in glycogen. What does this tell us about the mechanism of the reaction?
| 8.3.3 |
A hypothetical reaction mechanism of glycogen synthase |
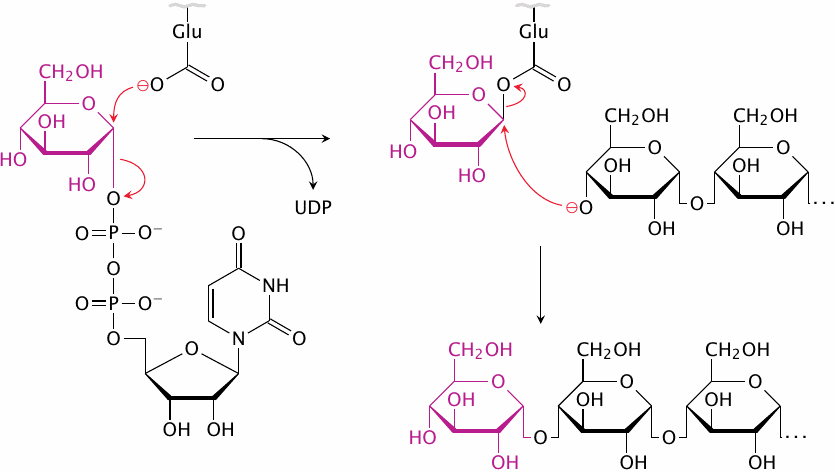
Nucleophilic substitutions can occur either synchronously or asynchronously. In the first case, which is called the SN2 mechanism, one substituent leaves as the other arrives; each of them holds on to the same carbon atom with “half a bond” in the transition state. Most commonly, the incoming substituent attacks from the direction opposite to the position of the leaving substituent. With asymmetric carbons such as the C1 of α-D-glucose, this should result in a reversal to the β configuration. The α configuration may then be restored in a second substitution.
The hypothetical scheme in this slide illustrates such a double SN2 mechanism. The first substitution leads to a covalent intermediate with an active site glutamate, in which the C1 of glucose has been inverted to the β configuration. The subsequent nucleophilic attack by the activated C4′ hydroxyl group restores the α form.
| 8.3.4 |
An alternative glycogen synthase mechanism |
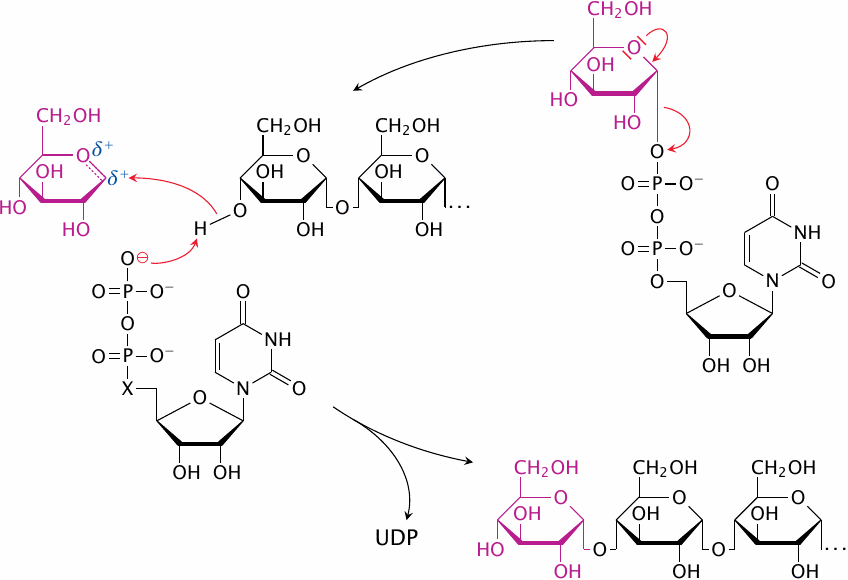
Double-substitution mechanisms similar to the one shown in the preceding slide are indeed employed by several glycosyltransferases, including glycogen phosphorylase and branching enzyme. However, with glycogen synthase itself, the evidence appears to favor a different mechanism that does not involve covalent catalysis by the enzyme, but instead a direct activation of both the C1 of the incoming glucose and the C4′ hydroxyl group of the preceding chain [34], as is depicted in simplified form in this slide. Note how the UDP that is released in the first step of the reaction assumes a catalytic role in the second step.
| 8.3.5 |
Overview of glycogen degradation |
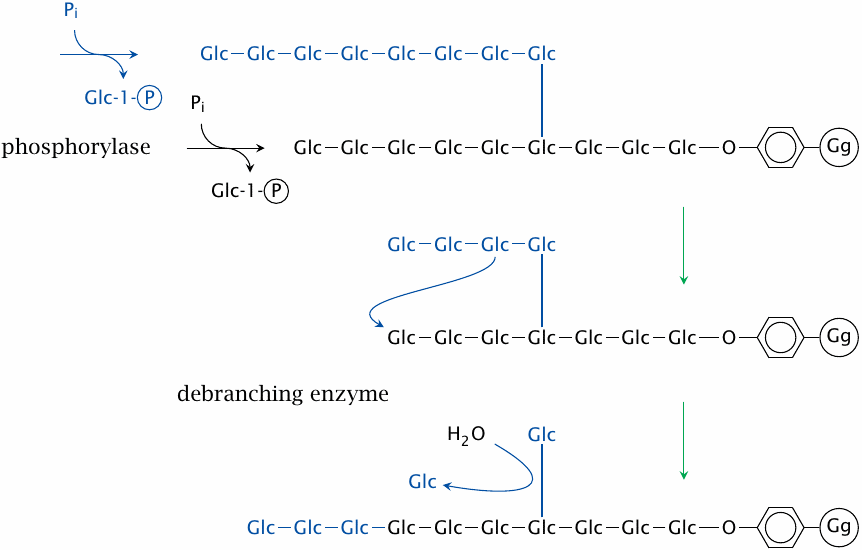
Glycogen degradation is brought about by phosphorylase and debranching enzyme. All glucose residues that are joined by α(1→4)-glycosidic bonds—that is, those in the straight segments—are released by glycogen phosphorylase. Most enzymes that cleave glycosidic bonds simply hydrolyze them; examples are intestinal amylase and β-galactosidase. In contrast, glycogen phosphorylase employs phosphate ions instead of water, and so produces glucose-1-phosphate rather than free glucose.48 Glucose-1-phosphate is then converted to the mainstream metabolite glucose-6-phosphate by phosphoglucomutase.
In the liver, which stores glycogen for the benefit of the entire body, the lion’s share of glucose-6-phosphate will be dephosphorylated by glucose-6-phosphatase and then released into the circulation; overall, this is equivalent to outright hydrolysis. However, muscle uses glycogen largely toward its own energy needs, and therefore glucose-6-phosphate will usually be funneled straight into glycolysis. In this case, the use of phosphorolysis instead of hydrolysis bypasses the hexokinase reaction, thereby saving one equivalent of ATP.49
Glycogen phosphorylase only degrades the chain ends to within four residues of a branching point. Then, debranching enzyme takes over and transplants the stub to another free end, where it becomes again a substrate for phosphorylase. However, this reaction leaves behind a single residue attached by a α(1→6)-glycosidic bond. This residue is subsequently released by the same enzyme as free glucose through hydrolysis.
| 8.3.6 |
The reaction mechanism of phosphorylase |
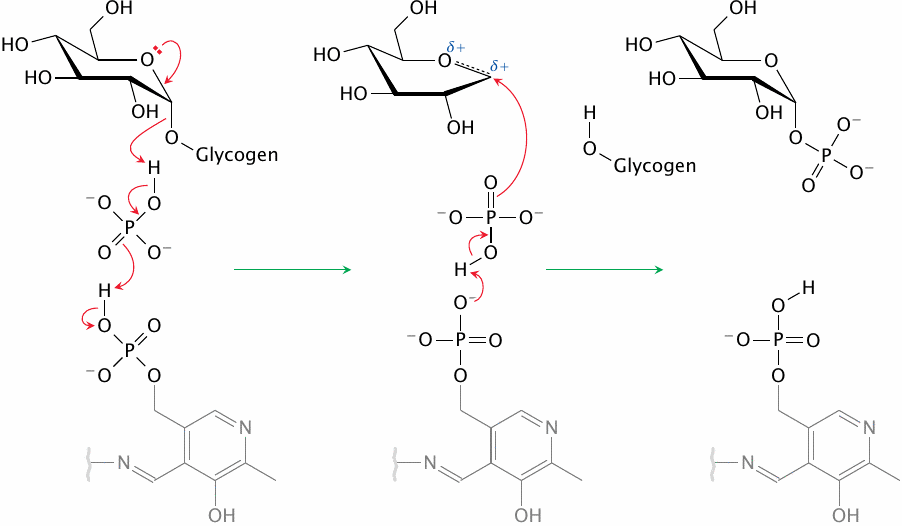
Glycogen phosphorylase uses pyridoxal phosphate (PLP) as a coenzyme. If you are familiar with the usual catalytic role of PLP, you may find this surprising; and indeed, phosphorylase employs it PLP a unique manner that bears no resemblance to its typical role in amino acid metabolism (see for example section 12.2). In those reactions, the organic ring of PLP, which is grayed out in this figure, functions as a reversible “electron sink.” In contrast, glycogen phosphorylase uses the phosphate group of PLP, which it deploys for acid-base catalysis to prime a free phosphate ion for attack on the terminal glycosidic bond of the glycogen substrate [35].
| 8.3.7 |
Lysosomal glycogen disposal |
- concerns a minor fraction of glycogen
- key enzyme: acid maltase; enzyme defect causes slow but inexorable glycogen accumulation
- possible role: disposal of structurally aberrant glycogen particles that have become “tangled up” during repeated cycles of glucose accretion and depletion
In liver cells, approximately 10% of all glycogen particles are found inside lysosomes [33], where they undergo slow degradation by acid maltase. This enzyme catalyzes the same reactions as intestinal amylase and maltase (see slide 1.6.12) but has an acidic pH optimum, in keeping with the acidic environment inside lysosomes (pH ~4.5). The lysosomal degradation pathway is important for the disposal of structurally aberrant glycogen particles; additional metabolic roles may exist but are currently not well understood. An enzyme defect for lysosomal maltase causes Pompe’s disease (slide 8.6.2).
| 8.4 |
Regulation of glycogen metabolism |
We have seen in slide 7.5.4 that phosphofructokinase and the complementary enzyme fructose-1,6-bisphosphatase are regulated by both intracellular and extracellular signals. The same applies to the key enzymes in glycogen metabolism.
| 8.4.1 |
Allosteric regulation of glycogen synthase and phosphorylase |
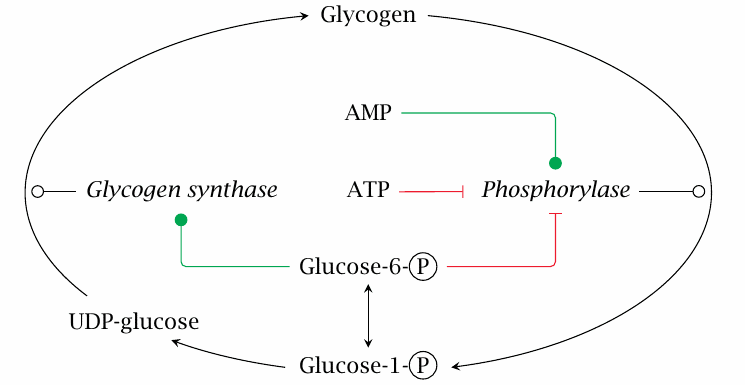
The allosteric regulatory effects exercised by glucose-6-phosphate, ATP and AMP on glycogen phosphorylase and glycogen synthase make good physiological sense. Depletion of ATP is an excellent reason to release glucose from the store in order to make some more. On the other hand, glucose-6-phosphate will be plentiful when glucose itself is abundant, and therefore signals an opportunity for replenishing the glycogen stores.
| 8.4.2 |
Hormonal control of glycogen metabolism |
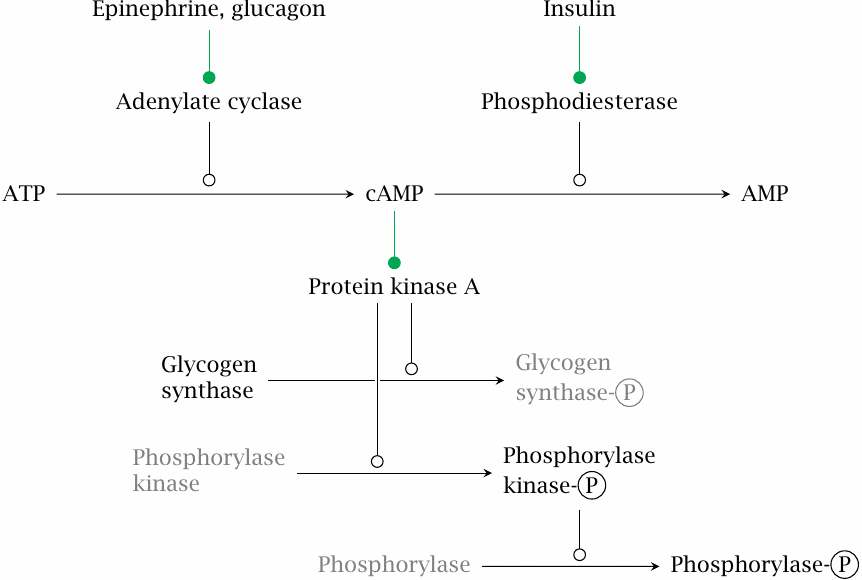
Hormonal control of glycogen metabolism is similar to that of gluconeogenesis; the cascade shown here is identical to that shown in slide 7.5.4 all the way from the hormones to the activation of protein kinase A. The activated kinase directly phosphorylates glycogen synthase, which inactivates that enzyme. Protein kinase A indirectly stimulates glycogen breakdown by phosphorylation of a dedicated regulatory enzyme, phosphorylase kinase, which in turn phosphorylates glycogen phosphorylase.
Note that glycogen synthase and phosphorylase respond in opposite ways to phosphorylation: The synthase is inactivated, whereas the phosphorylase is activated.
| 8.4.3 |
Regulatory differences between liver and muscle phosphorylase |
| Liver enzyme | Muscle enzyme | |
| Inhibition by glucose | + | − |
| Activation by Ca2+ | − | + |
| Activation by AMP even when unphosphorylated | − | + |
There are regulatory differences between glycogen phosphorylase in muscle and liver. Glucose inhibits the liver enzyme but not the muscle enzyme, and Ca2+ stimulates the muscle enzyme but not the liver enzyme. Recall that Ca2+ is also the trigger for muscle contraction; the simultaneous stimulation of glycogen breakdown therefore anticipates an increased demand for ATP.
As one would expect from their regulatory differences, the phosphorylases in liver and muscle are different molecules. Enzymes that catalyze the same reaction yet are separate molecules are referred to as isozymes. Although we usually don’t mention it, many other enzymes covered in this text occur as multiple isozymes, too.
| 8.5 |
Interorgan relationships in glycogen metabolism |
As stated above, the two tissues that have the most significant pools of glycogen are the liver and skeletal muscle. Liver glycogen is turned over rapidly; it serves as the major reserve of blood glucose during short-term fasts. Once liver glycogen is depleted, muscle glycogen can be drawn down; this, however, requires some roundabout metabolic trickery.
| 8.5.1 |
Liver glycogen utilization |
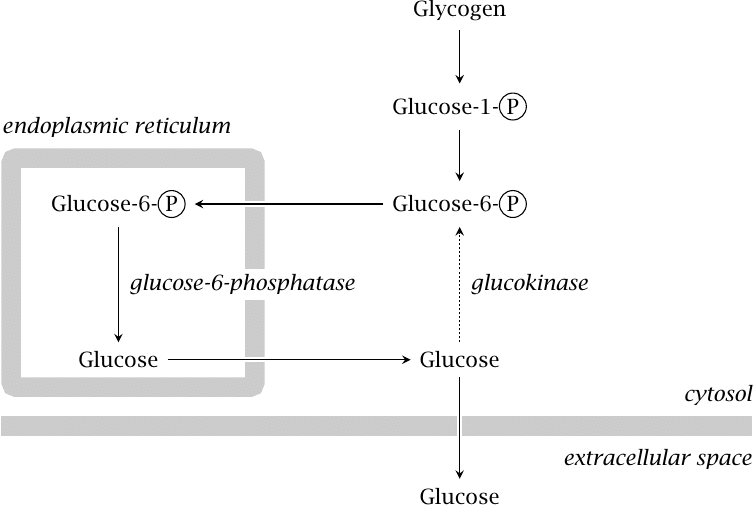
The liver mobilizes glucose from its glycogen store via glycogen phosphorylase and phosphoglucomutase, which yields glucose-6-phosphate. The latter is transported to the endoplasmic reticulum, where it is dephosphorylated. Glucose is taken back to the cytosol and released into the bloodstream.
Some of the glucose will be rephosphorylated before making it out of the cell, creating the futile cycle discussed in slide 7.5.2. However, the dominant glucose phosphorylating enzyme in the liver is glucokinase, which has fairly low affinity for glucose (see slide 3.5.3); therefore, enough glucose will escape rephosphorylation and be released into the bloodstream.
| 8.5.2 |
Muscle glycogen utilization |
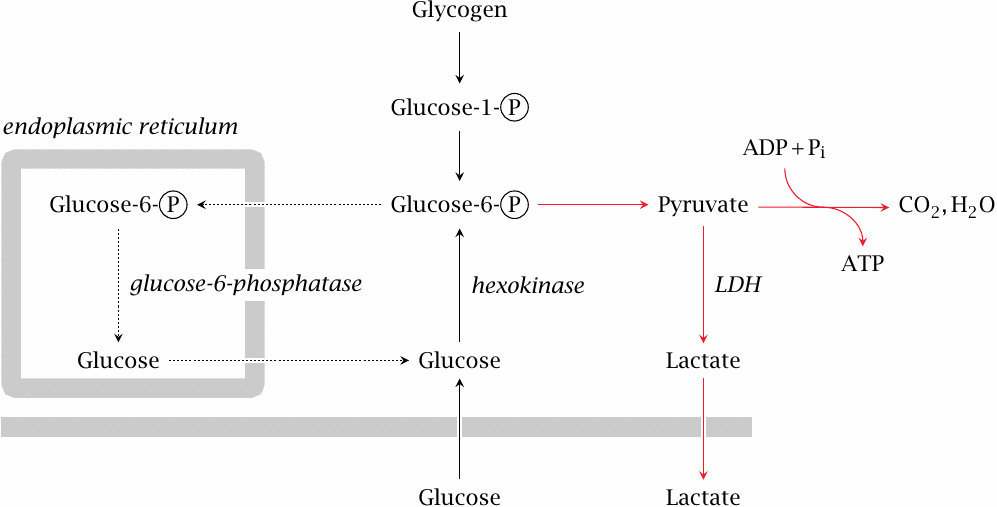
Muscle glycogen primarily serves the energy needs of muscle tissue itself; during prolonged physical exercise, most of it is broken down to glucose-6-phosphate and then consumed via the usual pathways right within the cells that stored it. As discussed above, this usage is facilitated by calcium-mediated activation of glycogen phosphorylase.
Under suitable conditions, namely, prolonged fast without physical exercise, muscle glycogen can also contribute to the replenishment of blood glucose. However, even though muscle cells have been shown to express glucose-6-phosphatase [36] and thus are, in principle, able to produce free glucose, they should find it difficult to release it. This is because muscle contains hexokinase, which has a much greater substrate affinity than glucokinase and therefore will keep the intracellular level of free glucose well below the extracellular concentration. The net transport of glucose should therefore be directed inward at all times; this agrees with all the physiological evidence that I could find.
The way around this obstacle is to convert glucose-6-phosphate to pyruvate and then lactate. At a low rate, lactate formation occurs even in resting muscle and under aerobic conditions. This lactate is derived in various proportions from blood glucose and glycogen, respectively. In animal experiments, epinephrine promotes glycogen utilization and lactate release [37–39], but overall the hormonal control of this process and the magnitude of its contribution to systemic glucose control are not well characterized.
| 8.5.3 |
The Cori cycle |
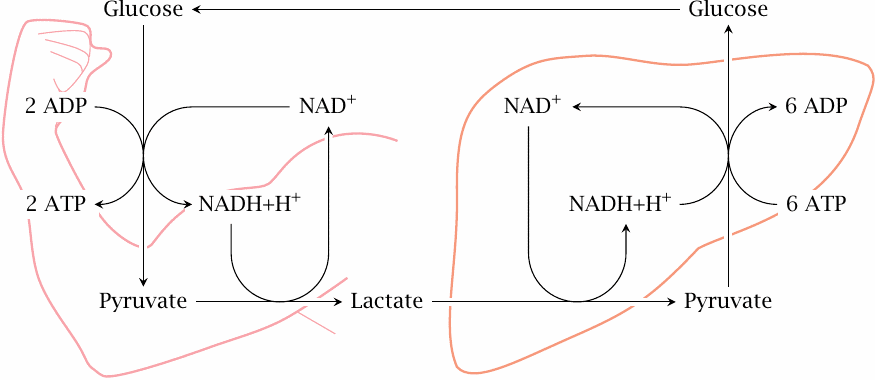
While skeletal muscle relies on oxidative metabolism most of the time, some other tissues, notably red blood cells and lymphocytes, which collectively account for some 4 kg of cell mass, depend mostly or even exclusively on anaerobic glycolysis even under aerobic conditions. The lactate released in peripheral tissues is scooped up by the liver, which converts it back to glucose through gluconeogenesis. This process is known as the Cori cycle, named after its discoverers Carl and Gerti Cori, who worked it out as early as 1929 [40].
Skeletal produces lactate at a very much higher rate during short bouts of maximal exercise when the ATP demand exceeds the capacity for aerobic metabolism. Some textbooks state that the Cori cycle resupplies the muscle with glucose in this situation also. This is, however, quite impossible. During intense exercise, the cardiac blood output is diverted from the visceral organs to skeletal muscle. Therefore, when ATP demand exceeds the oxygen supply of skeletal muscle, the oxygen shortfall would be even greater in the liver, should it indeed attempt to make enough ATP for sustaining the muscle through gluconeogenesis; and even with sufficient oxygen, its capacity for making glucose would fall far short of the muscles’ voracious appetite.
Anaerobic exercise can be sustained for only short periods of time anyway. During this period, the lactate turned out by skeletal muscle will simply accumulate; it will then slowly be scooped up by the liver and turned back into glucose after we have collapsed at the side of the track to catch our breath.
| 8.6 |
Glycogen storage diseases |
Genetic defects have been described for several enzymes of glycogen metabolism. The clinical syndromes associated with these defects are referred to as glycogen storage diseases. While these conditions are not particularly common, they do shed some light on the physiological significance of glycogen metabolism. Some conditions are clinically severe and are the focus of ongoing therapeutic research. A few examples are briefly discussed below.
| 8.6.1 |
Glucose-6-phosphatase deficiency (von Gierke disease) |
Biochemical defect:
- glucose-6-phosphate formed in gluconeogenesis or glycogen degradation cannot be converted to free glucose
- glucose cannot be exported from liver and kidney cells
Clinical manifestations:
- glycogen builds up in liver and kidneys (organ enlargement and functional impairment)
- severe hypoglycemia
- lactic acidosis
- hyperlipidemia
- hyperuricemia
Gluconeogenesis and glycogen degradation in liver and kidneys produce glucose-6-phosphate, which must then be dephosphorylated to glucose in order to be exported into the bloodstream (see slide 8.5.1). An enzyme defect for glucose-6-phosphatase prevents glucose release, which causes abnormally low blood glucose levels (hypoglycemia). Some of the surplus glucose-6-phosphate is funnelled into glycogen synthesis, whereas the remainder is converted to pyruvate in glycolysis and either emerges as lactate or, downstream of pyruvate dehydrogenase, is turned into triacylglycerol and cholesterol; the excess lactate and lipids account for the clinically observed lactic acidosis and hyperlipidemia, respectively.
The causation of hyperuricemia—excess blood levels of uric acid, see section 16.5—is less obvious. During episodes of hypoglycemia, the liver will be intensely stimulated by glucagon and epinephrine and make a forceful but futile attempt to mobilize its stored glycogen. The large amount of glucose-6-phosphate produced in this attempt, which cannot be converted to glucose, ties up and depletes cellular phosphate. This impedes the regeneration of ATP and raises the level of AMP, some of which then enters degradation to uric acid [41].50
The clinical severity of this disease may vary, presumably due to different levels of residual enzyme activity. Some cases may be managed with a diet of frequent, starch-rich meals, which helps to avoid hypoglycemia. In more severe cases, liver transplantation may become necessary.
| 8.6.2 |
Acid maltase deficiency (Pompe disease) |
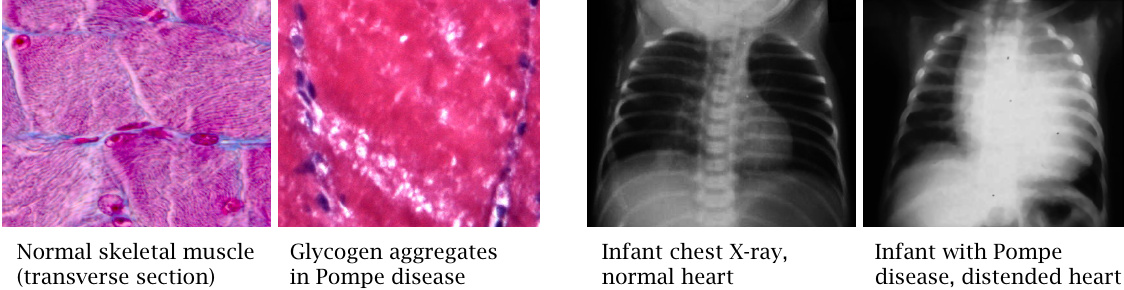
A homozygous deficiency of acid maltase51 disrupts lysosomal glycogen degradation and results in glycogen accumulation. Skeletal and heart muscle are more strongly affected than the liver. The tissue section of diseased muscle tissue shows “white holes,” which represent unstained aggregates of glycogen particles. Glycogen accumulation interferes with muscle cell function and contraction, and heart failure—a heavily impacted, severely distended heart is shown here in an X-ray image—leads to death.
The condition, which is known as Pompe’s disease, can vary in severity; complete lack of enzyme activity becomes manifest in infants, whereas mutations that reduce but do not completely inactivate the enzyme will cause milder disease with onset deferred to later childhood or adolescence. The disease can be treated with enzyme replacement therapy. The recombinant enzyme preparation is FDA-approved; its price is astronomical. This therapeutic approach is more thoroughly discussed in section 20.3.
While the nature of the enzyme defect would lead one to expect involvement of the interior organs (liver and kidney) also, these don’t seem to be prominent in practice, although some degree of hepatomegaly (liver enlargement) is often observed. I have not yet found a clear explanation for the preferential affliction of muscle tissues.
| 8.6.3 |
Muscle phosphorylase deficiency (McArdle’s disease) |
- Deficient glycogen breakdown inhibits rapid ATP replenishment
- Patients experience rapid exhaustion and muscle pain during exertion
- Liver phosphorylase and blood glucose homeostasis remain intact
Since liver and muscle phosphorylase are distinct isozymes, defects usually affect one and spare the other.52 In McArdle’s disease, the muscle isoform is selectively affected. An unexplained symptom in this disease is the so-called “second wind” phenomenon: during physical activity, patients initially fatigue rapidly, but then recover to a degree under continued exercise. This effect has been ascribed to the activation of protein and amino acid breakdown in muscle [43].
The lack of muscle phosphorylase should also inhibit the utilization of muscle glycogen toward blood glucose stabilization by way of the Cori cycle (see slide 8.5.3); one might therefore expect that McArdle’s disease might involve episodes of hypoglycemia. Interestingly, however, the literature does not contain reports of hypoglycemia in these patients.
| 8.6.4 |
Lafora disease |
- deficiency for laforin, a glycogen phosphatase
- accumulation of hyper-phosphorylated glycogen (Lafora bodies)
- patients develop epilepsy, dementia
Laforin is a phosphatase that is associated with glycogen particles and removes phosphate groups from glycogen itself [44]. The functional significance of glycogen phosphorylation and dephosphorylation is not clear; and you will notice that it was not even mentioned above. However, genetic deficiencies of the phosphatase lead to the accumulation of Lafora bodies, which consist of phosphorylated, poorly branched glycogen molecules. The disease becomes manifest through a specific form of epilepsy (myoclonic seizures) and dementia and is fatal. The CNS symptoms—like the involvement of the kidneys in v. Gierke disease, see above—illustrate that tissues other than liver and muscle contain glycogen as well and may be damaged by its accumulation.