| 11 |
Cholesterol metabolism |
| 11.1 |
Biological significance of cholesterol |
- Cholesterol is an essential lipid constituent of cell membranes
- Cholesterol is a precursor of steroid hormones and of bile acids
- Intermediates of cholesterol biosynthesis are required to make vitamin D and for posttranslational modification of membrane proteins
- High plasma cholesterol promotes atherosclerosis
Contrary to popular belief, the biological role of cholesterol is not limited to being the bad guy. Instead, it has a number of essential physiological functions:
- 1.In humans and animals, cholesterol is a major constituent of the cell membranes. Cholesterol modulates physical properties of these membranes that in turn affect the function of membrane proteins such as receptors and transporters. Experimental depletion of membrane cholesterol cripples many cellular functions.
- 2.Cholesterol is the biosynthetic precursor of bile acids, which are essential for fat digestion.
- 3.Cholesterol is the precursor of all steroid hormones, namely, androgens, estrogens, progestins, glucocorticoids, and mineralocorticoids, as well as of calciferol (vitamin D).
Nevertheless, it certainly is true that cholesterol also plays a major role in the pathogenesis of atherosclerosis. In this chapter, we will first cover the metabolic pathways that involve cholesterol, and then take a look at the role of cholesterol in this important cardiovascular disease.
| 11.1.1 |
Processes that determine the cholesterol balance |
- intestinal uptake of dietary cholesterol
- de novo cholesterol synthesis
- synthesis of steroid hormones from cholesterol
- synthesis of bile acids from cholesterol, and their biliary secretion
- biliary secretion of surplus cholesterol in unmodified form
Cholesterol can both be synthesized endogenously and obtained from the diet. Note that significant amounts of cholesterol only occur in meat, eggs, and milk products; plants and mushrooms contain other sterols but very little cholesterol. The observation that vegetarians survive tells us that our capacity to synthesize cholesterol suffices to cover our need for cholesterol entirely.
| 11.2 |
Cholesterol synthesis |
The pathway of cholesterol synthesis is quite elaborate. We will start with a general outline and then go through most, but not all of the reactions in detail.
| 11.2.1 |
Overview of cholesterol synthesis |
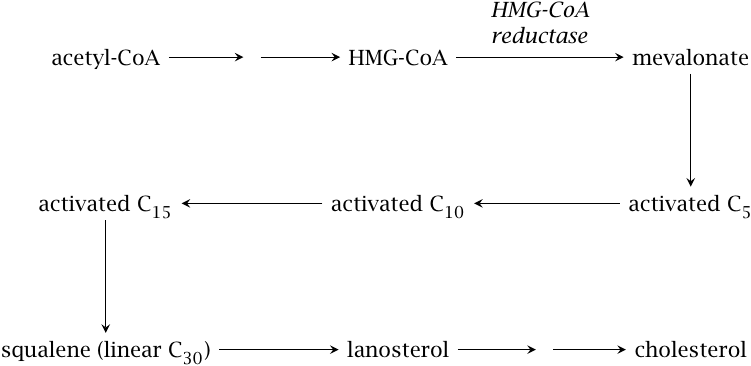
Cholesterol synthesis starts with acetyl-CoA, which is used to synthesize hydroxymethylglutaryl-CoA (HMG-CoA). The reactions in this initial stage are the same as in ketogenesis (see slide 10.4.1). However, while ketogenesis occurs in the mitochondria, HMG-CoA destined for sterol synthesis is formed in the cytosol. Therefore, like the synthesis of fatty acids, cholesterol biosynthesis depends on the export of acetyl-CoA from the mitochondria. Also as with fatty acids, multiple steps in the cholesterol synthesis require NADPH. How these two requirements are met has been discussed earlier (see Section 10.5.6f and Chapter 9).
All steps downstream of HMG-CoA occur in the smooth endoplasmic reticulum. HMG-CoA reductase reduces HMG-CoA to mevalonate; this enzyme is the major target of regulation in the entire pathway. Mevalonate is converted to various isoprene intermediates. Several rounds of “polymerization”—I’m using the term loosely—produce the linear hydrocarbon molecule squalene, which is cyclized to the first sterol intermediate. This molecule, lanosterol, is then converted to cholesterol by several successive modifications.
| 11.2.2 |
Initial activation steps in cholesterol synthesis |
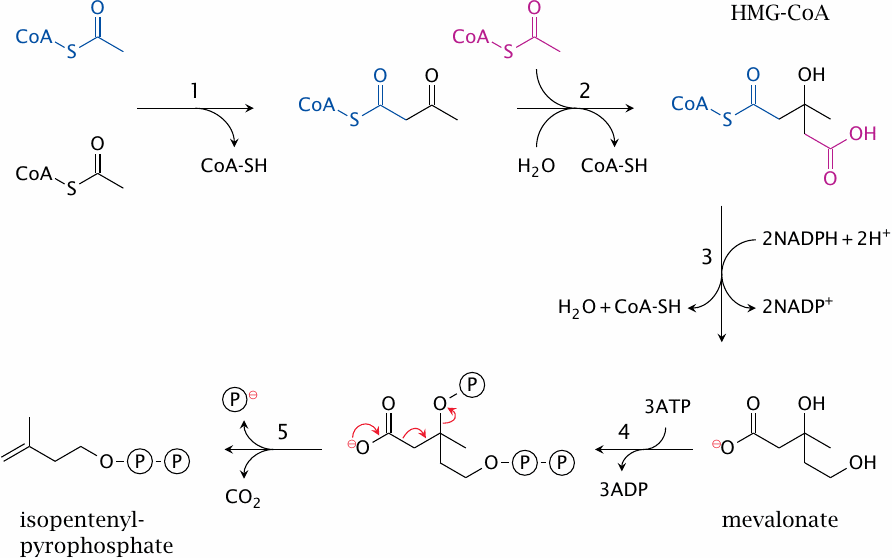
The reactions shown in this slide are catalyzed by thiolase (1), HMG-CoA synthase (2), HMG-CoA reductase (3), mevalonate kinase, phosphomevalonate kinase (4), and diphosphomevalonate decarboxylase, and diphosphomevalonate decarboxylase again (5).68 In the subsequent steps of the pathway, six molecules of isopentenyl-pyrophosphate are used for the synthesis of one cholesterol molecule.
| 11.2.3 |
Formation of a C10 intermediate |
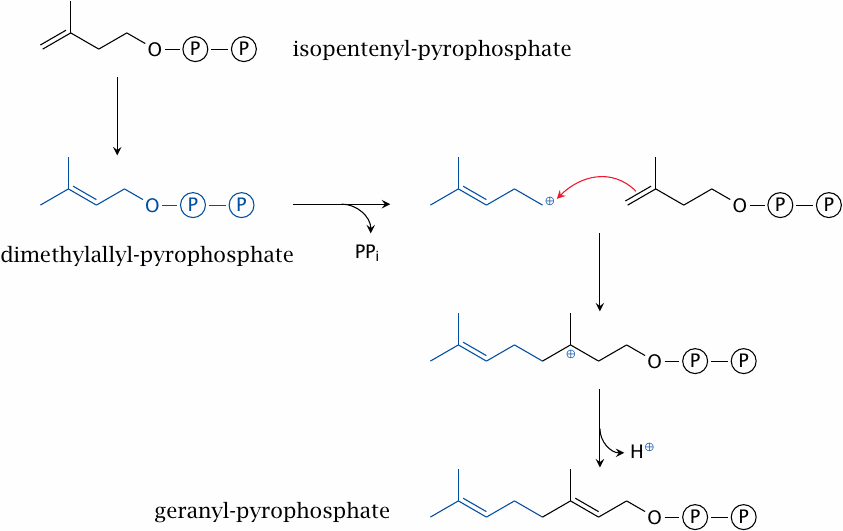
The next stage begins with the conversion of one molecule of isopentenyl-pyrophosphate to dimethylallyl-pyrophosphate, catalyzed by the isopentenyl-pyrophosphate isomerase. The product is condensed with another molecule of isopentenyl-pyrophosphate to yield geranyl-pyrophosphate. In this reaction, catalyzed by geranyl-pyrophosphate synthase, the pyrophosphate of the first substrate serves as a leaving group. The resulting carbocation reacts with the double bond of the second substrate.
| 11.2.4 |
Formation of C15 and C30 intermediates |
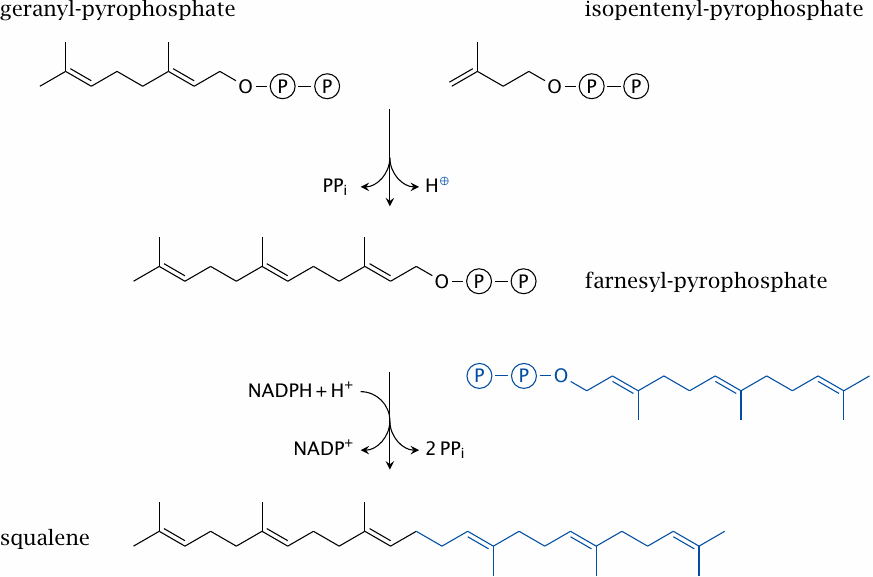
The synthesis of the C15 intermediate, farnesyl-pyrophosphate, is catalyzed by the eponymous synthase and mechanistically resembles that of geranyl-pyrophosphate. Farnesyl-pyrophosphate is used not only in sterol synthesis but also in the posttranslational modification of some membrane-associated proteins. While the amount of farnesyl-pyrophosphate used for the latter purpose is not very large, inhibition of protein farnesylation may contribute to the clinical effect of inhibitory drugs that act upstream in this pathway; this includes the statins, which inhibit HMG-CoA reductase (see slide 11.7.2).
Two molecules of farnesyl-pyrophosphate are joined head to head in the synthesis of the final linear sterol precursor, namely, squalene; the enzyme is named squalene synthase.
| 11.2.5 |
Squalene cyclization yields the first sterol intermediate |
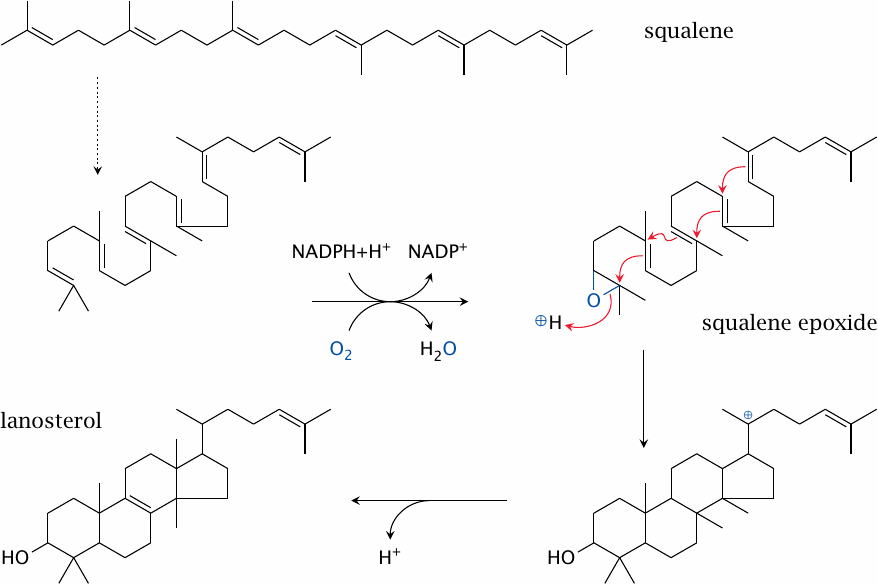
The reactions shown here are catalyzed by squalene epoxidase and lanosterol synthase. The rearrangement indicated by the dashed arrow is not a real reaction—we just rotate a couple of single bonds to show how the pieces fall into place for the subsequent cyclization.
The oxygen is introduced by squalene epoxidase, a cytochrome P450 enzyme. Such enzymes use NADPH to reduce one of the two atoms of molecular oxygen, while retaining the other one in a highly reactive state, which they then use toward their specific purposes (see slide 19.2). Squalene synthase inserts its active oxygen into a C=C double bond of the substrate to form an epoxide. The subsequent cleavage of the epoxide by lanosterol synthase starts a cascade of reactions that goes from one end of the molecule to the other, closing all four rings of the sterol skeleton in the process. Note that a methyl group also changes its place on the sterol ring; the reaction mechanism is quite intricate.
| 11.2.6 |
Demethylation, desaturation and saturation steps convert lanosterol to cholesterol |
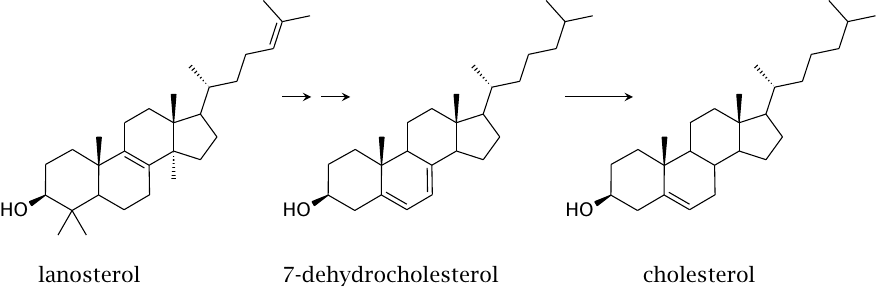
Several successive modifications convert lanosterol to 7-dehydrocholesterol and then cholesterol. Like squalene epoxidase, several of the enzymes that catalyze these reactions also belong to the cytochrome P450 family. We will not consider them in detail; however, several of these enzymes are inhibited by the drug triparanol, which was once used to treat hypercholesterolemia before being withdrawn due to toxicity.
| 11.2.7 |
UV-dependent synthesis of cholecalciferol |
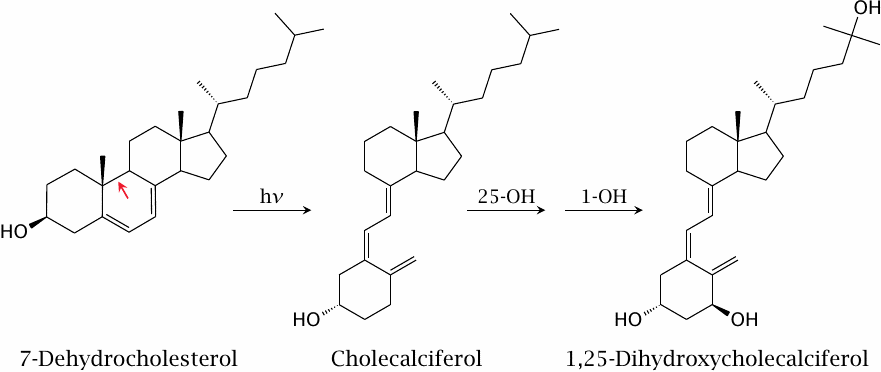
The last biosynthetic precursor of cholesterol, 7-dehydrocholesterol, is also the precursor of vitamin D3 (cholecalciferol). The conversion of 7-dehydrocholesterol to cholecalciferol, which involves the breakage of the bond indicated by the red arrow, is a photochemical reaction; it requires the absorption of a UV photon and can therefore only occur in the skin. Two successive enzymatic hydroxylations (which do not require any more UV light) yield 1,25-dihydroxy-cholecalciferol. This molecule is is a transcriptional regulator that activates the uptake of calcium and phosphate from the gut.
A lack of 1,25-dihydroxy-cholecalciferol causes rickets, a formerly quite common disease that is characterized by lack of bone mineral and bone deformities. Cholecalciferol is also the vitamin du jour, and if we are to believe the news, the deficiency also causes depression, cancer, earthquakes, and at least 105 % of all other evils befalling mankind.
The essential function of 1,25-dihydroxycalciferol is responsible for the variation of human skin colors. While dark pigment protects the skin from damage by UV irradiation, the UV photons swallowed up by the pigment are no longer available for the synthesis of cholecalciferol. When Homo sapiens left Africa’s sun for cloudier climates, the shortage of sunshine created a selective pressure for lighter skin, which increases the availability of photons for cholecalciferol synthesis. The remarkably pale skin of people of British or Irish descent tells you all you need to know about the weather in those places. Avoid.
| 11.2.8 |
Sterol metabolism occurs in the smooth endoplasmic reticulum |
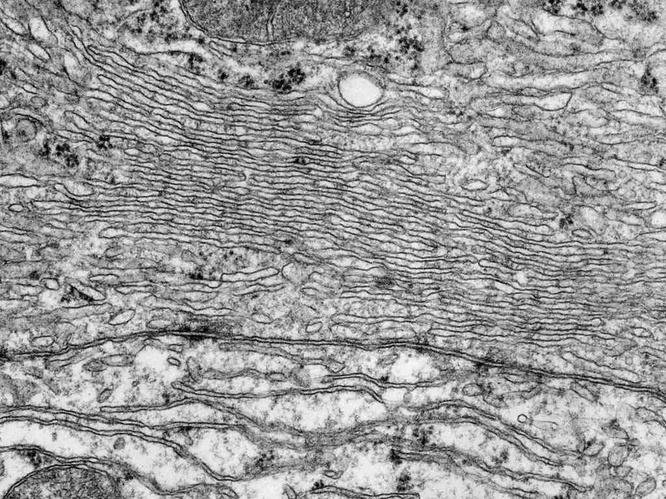
Many intermediates in the synthesis of cholesterol and of its derivatives are very hydrophobic and very poorly soluble in water; they therefore have to reside in an apolar environment. On the other hand, the enzymes, like almost all proteins, are at least partially polar and cannot immerse completely in the same apolar environment.
The solution to this problem is to perform the reactions at the interface of polar and apolar environments, that is, at membrane surfaces. This means that the membrane of the smooth ER is not just there to delimit a separate compartment—it is a reaction compartment of its own. Accordingly, liver cells and other cells that engage in sterol chemistry have a well developed smooth ER with a very large cumulative membrane surface area.
The ER also hosts many enzymes that function in drug metabolism, including cytochrome P450 and UDP-glucuronosyltransferases (see chapter 19). The functional context is the same—many drug molecules that must undergo metabolism before being eliminated are quite hydrophobic and thus also require interfacial chemistry.
| 11.3 |
Regulation of cholesterol synthesis |
HMG-CoA reductase subject to feedback inhibition by cholesterol. Additionally, its activity is subject to transcriptional regulation, which occurs through a rather unique mechanism.
| 11.3.1 |
Transcriptional regulation of cholesterol synthesis starts in the endoplasmic reticulum |
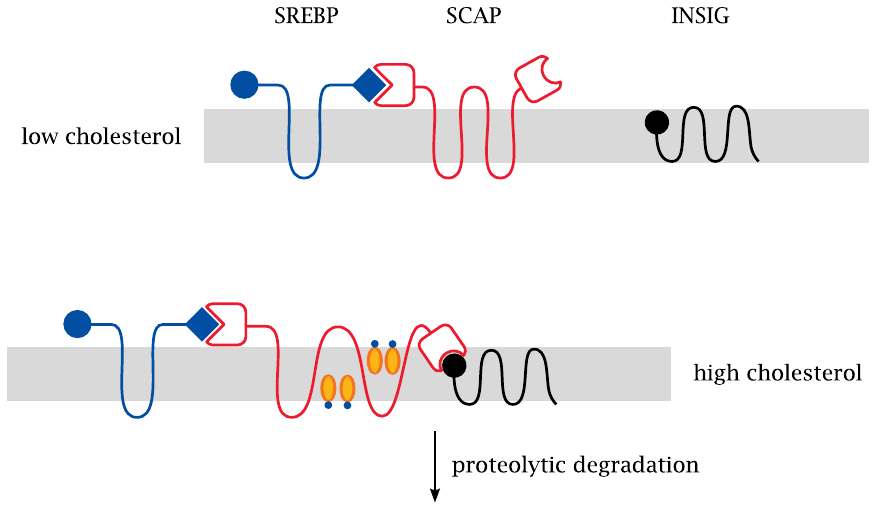
The sterol response element (SRE) is a DNA consensus sequence that controls the transcription of HMG-CoA reductase. The corresponding SRE-binding protein (SREBP) is initially embedded in the ER membrane, and thus evidently unable to get in touch with its DNA target. SREBP is bound to a second protein, namely, SREBP cleavage activating protein (SCAP). This protein is the actual cholesterol sensor; it can adopt two different conformations, depending on the content of cholesterol in the surrounding membrane. The conformation that predominates at high cholesterol content lets SCAP bind to a third protein, INSIG.69 When this ternary complex forms, it is rapidly targeted toward proteolytic degradation, and that is the end of it.
At low cholesterol concentrations, however, SCAP does not bind to INSIG, and this is when things get interesting, as shown in the next slide.
| 11.3.2 |
When cholesterol is low, SREBP is sorted to the Golgi apparatus |
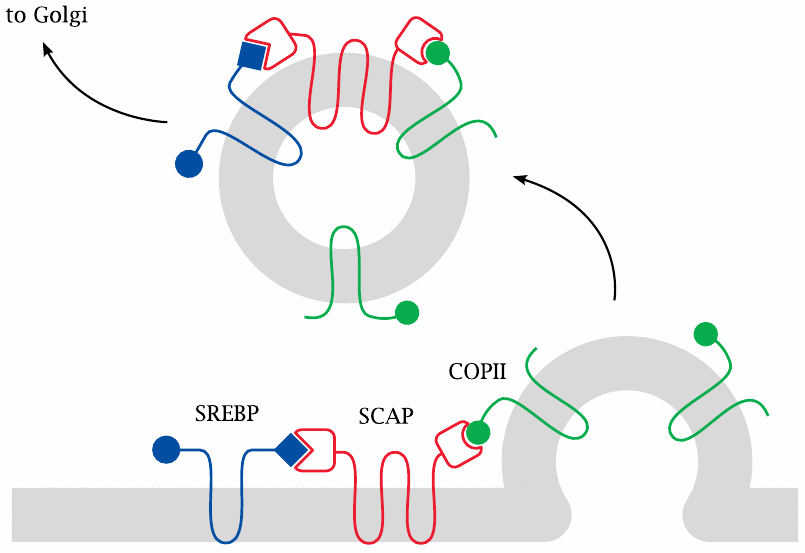
At low cholesterol concentrations, SCAP binds to another membrane protein (COPII, shown in green) that recruits it into nascent vesicles, and it takes SREBP along for the ride. When these vesicles bud off from the ER membrane, they travel to the Golgi apparatus and fuse with its membrane. The Golgi is a cellular organelle that performs many types of posttranslational protein modification, such as glycosylation, lipid modification, and proteolytic processing.
| 11.3.3 |
Proteolytic cleavage in the Golgi releases SREBP |
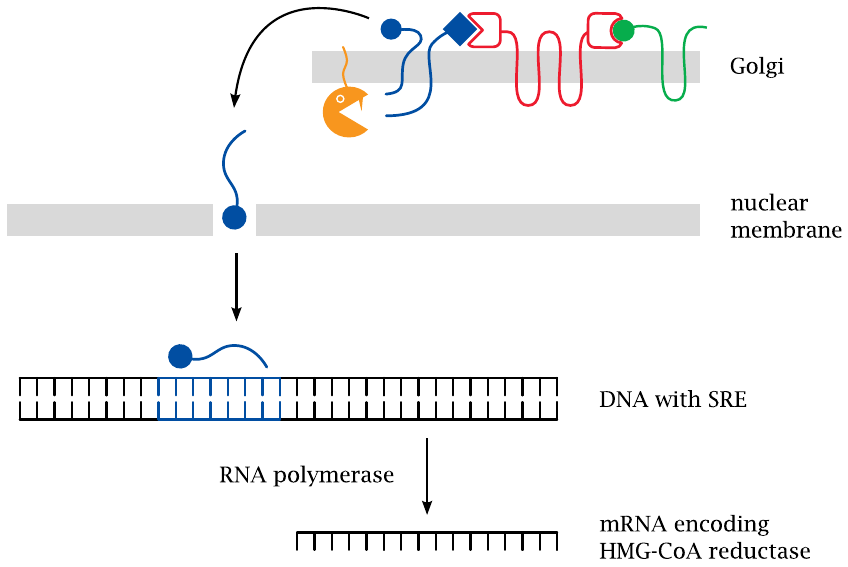
Once SREBP reaches the Golgi, it is ambushed and cleaved by two specific proteases (S1P and S2P). Cleavage releases the DNA-binding domain of the protein from the membrane. This fragment then moves across the cytosol and enters the nucleus, where it binds to SRE sequence elements that increase the expression of HMG-CoA reductase and also of various other enzymes from the cholesterol synthesis pathway [66].
Another protein that is upregulated by SREBP and SRE is the LDL receptor, a membrane protein that mediates endocytosis of low density lipoprotein (LDL; see slide 11.4.7). In cells that do not synthesize cholesterol themselves, SREBP upregulates transcription of the LDL receptor,
Like SREBP and SCAP, HMG-CoA reductase is anchored in the ER membrane. This does not seem necessary for the chemistry it performs. Instead, this location facilitates the negative feedback regulation imposed on it by cholesterol. Indeed, the enzyme contains a sterol-sensing domain that is homologous to the one found in SCAP [67].
| 11.4 |
Cholesterol transport |
Like other lipids, cholesterol has low water solubility and therefore requires special mechanisms and vehicles for transport. In the bloodstream, both cholesterol and triacylglycerol are transported within lipoproteins.
| 11.4.1 |
Lipoprotein structure |
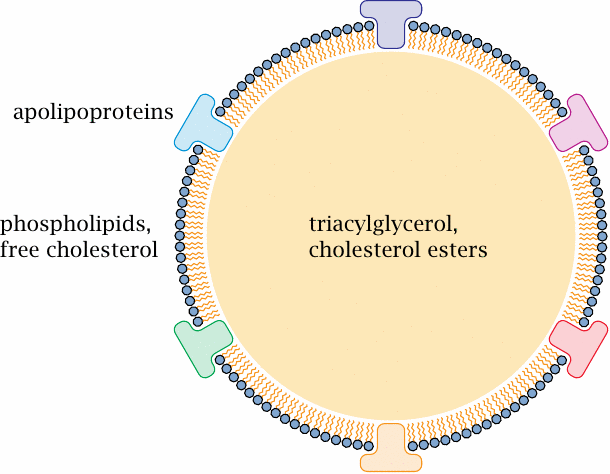
We had already encountered one type of lipoprotein, namely, the chylomicrons, which are formed in the intestinal mucosa (see slide 10.2.3). Different lipoprotein types vary in composition and particle size, but the overall structure is similar for all of them. The surface consists mainly of phospholipids, which form a monolayer.
Apolipoproteins are embedded into the surface of the particle. These protein molecules mainly serve as “address tags” that mediate the interaction with target molecules and cells, such as lipoprotein lipase on endothelial cells (slide 10.2.6) or the LDL receptor (see later). Some apolipoproteins are stably associated with their lipoprotein particles; for example, a given molecule of apolipoprotein B remains associated with the same chylomicron or VLDL (very low density lipoprotein, see below) particle throughout its lifetime. Exchangeable apolipoproteins have regulatory roles; for example, apolipoprotein CII reversibly associates with VLDL particles and promotes their interaction with lipoprotein lipase.
| 11.4.2 |
Classification of plasma lipoproteins |
| Chylomicrons | VLDL | LDL | HDL | |
| Density (g/ml) | 0.95 | 0.95–1.0 | 1.02–1.06 | 1.06–1.12 |
| Origin | small intestine | liver | liver | liver |
| Function | distribute dietary TAG and cholesterol | distribute TAG from liver | distribute cholesterol from liver | return excess cholesterol to liver |
| Predominant lipid species | TAG | TAG | cholesterol | phospholipids, cholesterol |
As you will notice, the variation in density between the various lipoproteins is modest, but it is large enough to allow their separation by density gradient centrifugation, and they are classified according to their behavior in this fractionation procedure. As noted before, ‘LDL’ stands for low density lipoprotein; ‘HDL’ means high density lipoprotein, and ‘VLDL’ is very low density lipoprotein. The differences in density arise from two circumstances:
- 1.Lipids are lighter than protein. Within the particles, proteins are found only at the surface, whereas the interior contains only lipids; therefore, the fractional content of protein is lower with larger particles than with smaller ones. Chylomicrons are the largest and least dense lipoprotein species.
- 2.Triacylglycerol is lighter, but cholesterol is heavier than water. Accordingly, a high content of cholesterol in the lipid fraction will also increase the overall density.
Note the central role of the liver, which orchestrates most of the lipid transport, with the exception of intestinal lipid uptake and packaging into chylomicrons. We will now first look at the uptake of cholesterol in the intestine and then at its transport to other organs.
| 11.4.3 |
Two membrane proteins control the uptake of sterols from the intestine |
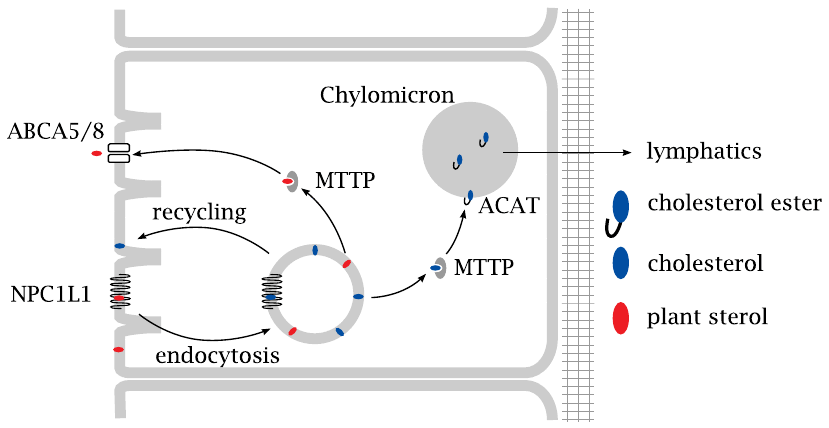
The uptake of cholesterol by intestinal epithelial cells begins with endocytosis. This process is controlled by NPC1L1, a membrane protein which is deficient in a lipid storage disorder known as Niemann-Pick disease. NPC1L1 is a sterol sensor and promotes cholesterol uptake through endocytosis.
From the endocytotic vesicles, cholesterol is transferred to the endoplasmic reticulum by the microsomal triglyceride transfer protein (MTTP). Acylation by acyl-CoA cholesterol acyltransferase (ACAT) yields a cholesterol ester, which is loaded into a nascent chylomicron together with triacylglycerol. After the chylomicrons have been released from the intestinal cells and reached the circulation via the lymphatics (see slide 10.2.5), most of their triacylglycerol is depleted by capillary lipoprotein lipase. The cholesterol stays behind in the chylomicron remnants, which are taken up and utilized in the liver (see slide 10.2.6).
In contrast to what its name suggests, MTTP transports not only triacylglycerol but also sterols. Mutational inactivation of this protein results in abetalipoproteinemia.70 Such patients have reduced levels of chylomicrons and are affected by malabsorption of lipids and of lipid-soluble vitamins.
The NPC1L1-mediated uptake of cholesterol by endocytosis does not discriminate between cholesterol and other, structurally similar sterols derived from plants. After uptake, the latter are diverted toward a transport protein in the apical membrane, ABCA5/8, which expels them right back into the gut lumen through active transport.
| 11.4.4 |
Plant sterol structures |
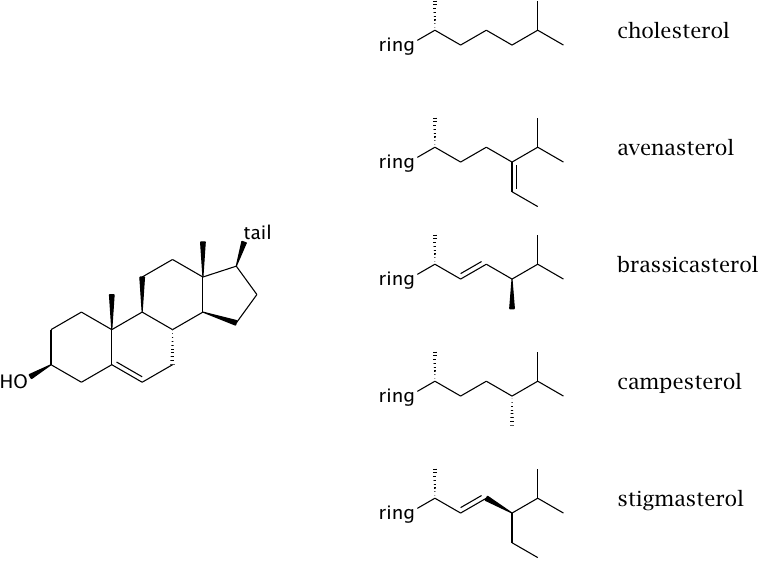
Plants contain very little cholesterol but instead contain a variety of structurally similar sterols. The sterol ring is the same as with cholesterol in all sterols shown, but the tails are somewhat different.
Plant sterols compete with cholesterol for “space” inside the cytoplasmic membrane of intestinal cells, and therefore reduce the rate of cholesterol absorption by endocytosis. Dietary application of sitosterol or other plant sterols is a moderately effective strategy to reduce cholesterol absorption.
| 11.4.5 |
Structures of ABC transporters in the inward-open and outward-open conformations |
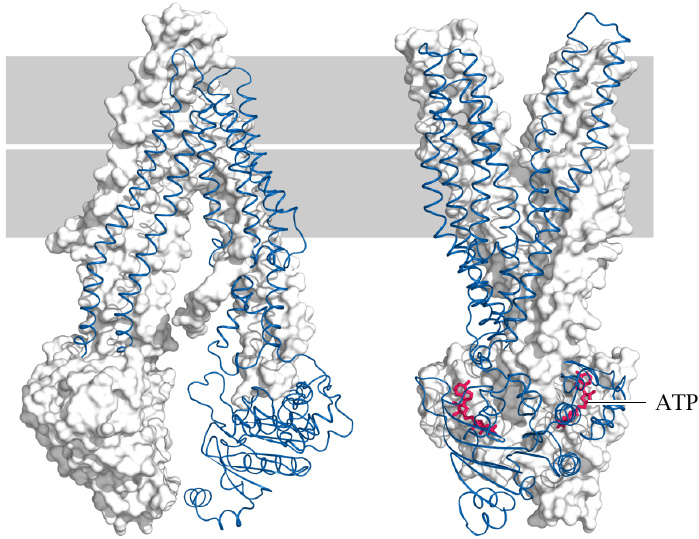
Both ABCA5/8 (previous slide) and ABCA1 (slide 11.4.7) are members of the ATP-binding cassette or ABC family of transporters. These have a common structural organization. Several ABC transporters have been crystallized in the inward- and outward open conformations [68,69], and the two structures provide a glimpse of how they work.
ABC transporters often have rather broad substrate specificity and mediate the membrane translocation of many metabolites and xenobiotics. In addition to cholesterol and other membrane lipids, important examples are bile acids (slide 11.5.3), conjugated bilirubin (slide 17.4), drugs, and drug metabolites (19.1.3). Cancer cells often overexpress ABC transporters, which renders them resistant to multiple anticancer drugs.
| 11.4.6 |
ABC transporters induce substrate “flip-flop” across the membrane |
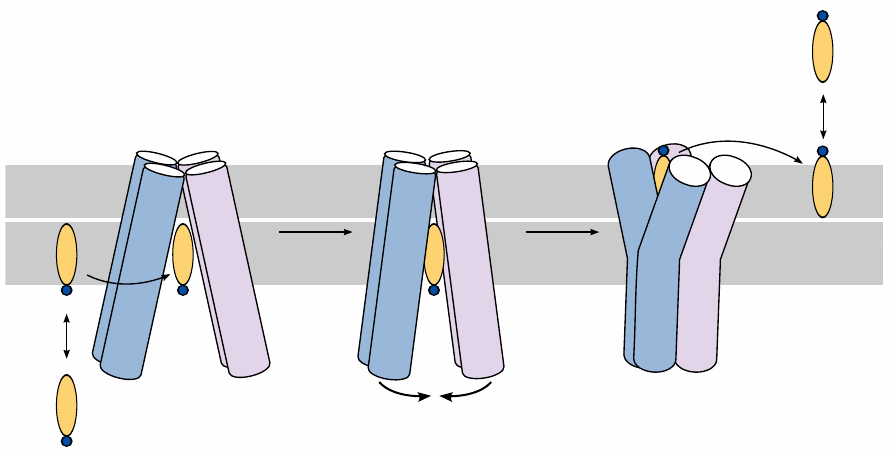
One feature that is shared by many ABC transporter substrates is their amphiphilic nature. Most ABC transporters expel their substrates from the cytosol to the extracellular space. In this case, the substrate initially resides within the inner leaflet of the cytoplasmic membrane. Once it enters the inward-open conformation of the transporter, the latter undergoes a transition to the outward-open conformation, which is powered by the hydrolysis of ATP. The substrate then leaves the transporter and diffuses into the outer membrane leaflet, from where it may distribute to other extracellular reservoirs.
| 11.4.7 |
Transport of cholesterol between the liver and peripheral tissues |
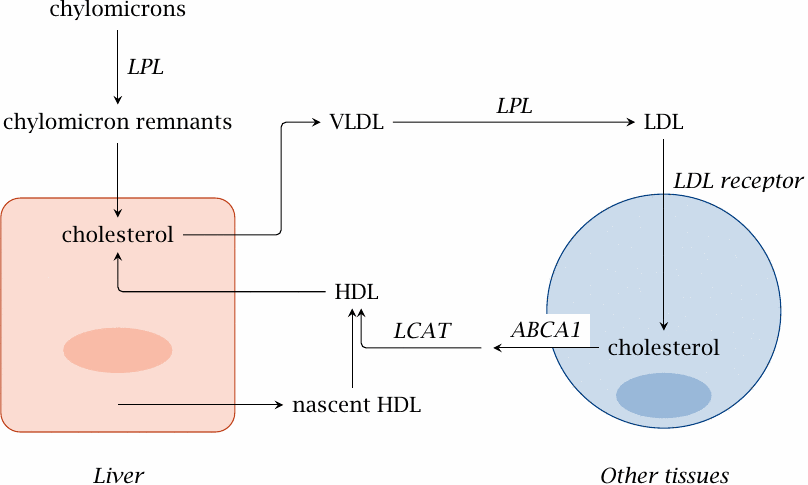
The liver synthesizes cholesterol from acetyl-CoA (section 11.2). The cholesterol pool in liver cells also receives the dietary cholesterol, which is contained in the chylomicron remnants that are formed through the extraction of triacylglycerol from chylomicrons by lipoprotein lipase (LPL; slide 10.2.6).
Liver cells package esterified cholesterol, together with triacylglycerol, into particles of very low density lipoprotein (VLDL). Like chylomicrons, VLDL interacts with lipoprotein lipase and thereby turns into intermediate (IDL) and then low density lipoprotein (LDL).
LDL is taken up by cells in the periphery through endocytosis, which is mediated by the LDL receptor.71 Excess cholesterol is exported from the cell by an active transporter (ABCA1) and delivered to high density lipoprotein (HDL), which then carries it back to the liver. Cholesterol transport by HDL is facilitated by lecithin-cholesterol acyltransferase (see next slide).
| 11.4.8 |
The lecithin-cholesterol acyltransferase (LCAT) reaction |
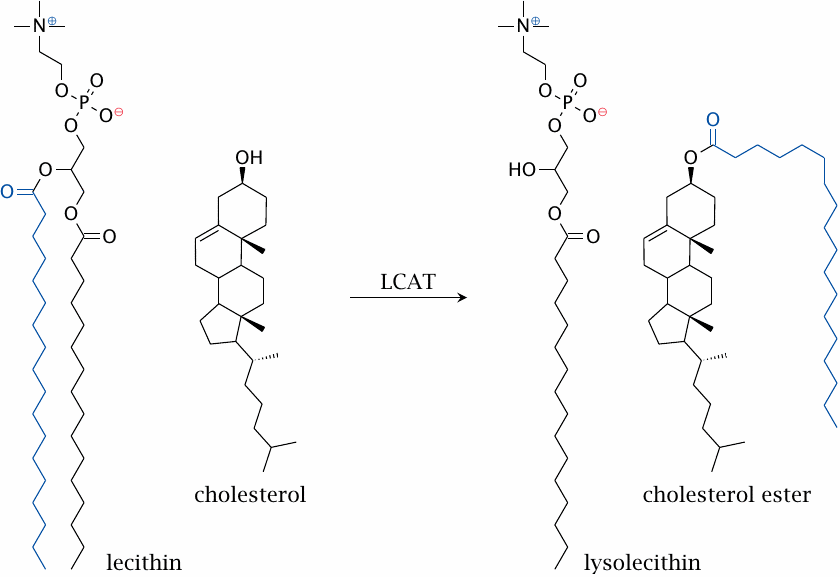
The HDL particle contains the enzyme lecithin cholesterol acyltransferase, or LCAT for short, which converts cholesterol to cholesterol esters.
The LCAT reaction occurs at the surface of HDL particles. The transfer of one acyl chain from a lecithin (phosphatidylcholine) molecule to cholesterol produces a cholesterol ester and lysolecithin. The significance for cholesterol transport is illustrated in the next slide.
Cholesterol also undergoes esterification as it is packaged into chylomicrons and VLDL inside intestinal and liver cells, respectively. In these cases, acyl-CoA serves as the donor of the acyl residue (see slide 11.4.3).
| 11.4.9 |
Cholesterol esters can be stored inside lipoprotein particles |
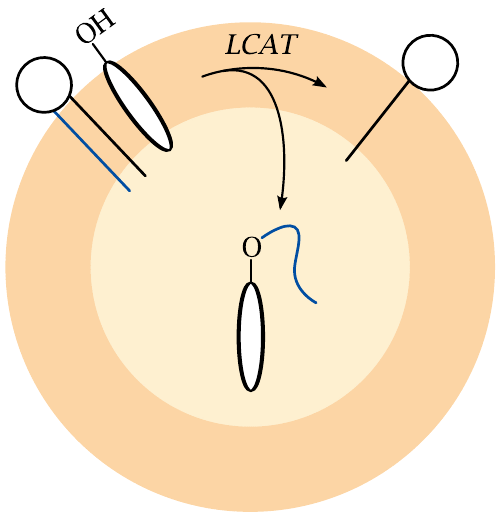
Cholesterol is amphiphilic and tends to accumulate at lipid/water interfaces, with the OH group exposed to the aqueous phase. Free cholesterol can therefore be transported only within the outermost layer of lipid molecules of a lipoprotein particle. In contrast, cholesterol esters are entirely hydrophobic and readily partition into the interior of lipoprotein particles. The LCAT reaction therefore greatly increases the transport capacity of HDL particles for cholesterol.
| 11.5 |
Bile acid metabolism and transport |
Bile acids are the quantitatively most important derivatives of cholesterol. Aside from their essential role in fat digestion, they are also required to keep hydrophobic constituents of the bile in solution, such as unconjugated bilirubin (see section 17.4) and cholesterol itself.
| 11.5.1 |
Bile acids are derived from cholesterol |
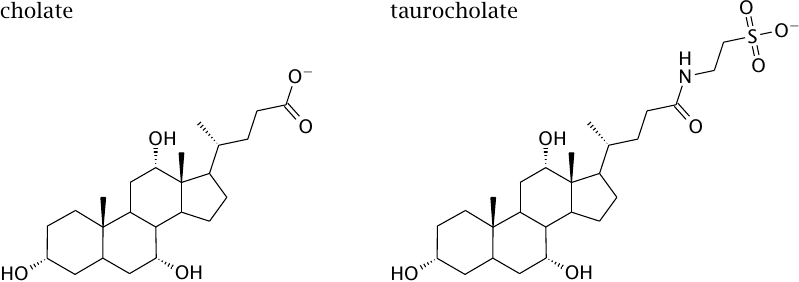
Cholic acid and some other bile acids are synthesized from cholesterol in the liver. Taurocholate is produced through conjugation of cholate with taurine; similarly, glycocholate is produced through conjugation with glycine.
| 11.5.2 |
Bile acids undergo enterohepatic cycling |
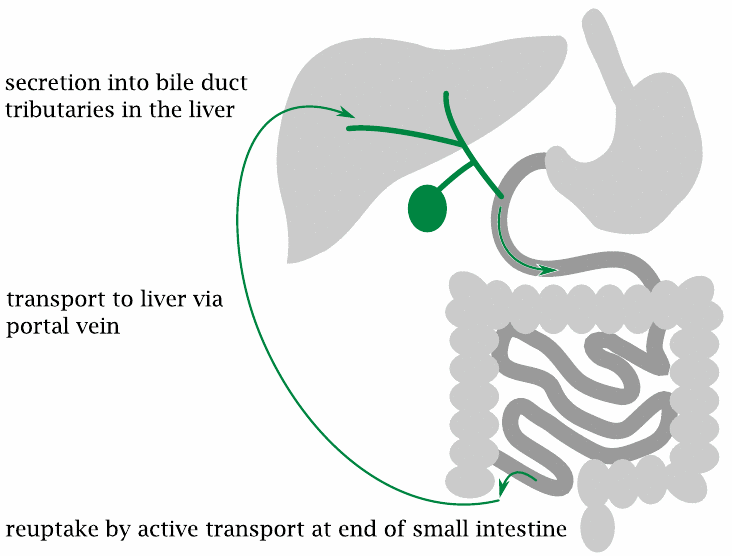
In an enterohepatic cycle, a substance is secreted by the liver into the bile, passes into the intestine and is taken up again into the blood, either by passive diffusion across cell membranes or by active transport. Since blood drained from the intestines feeds into the portal vein, the substance will return to the liver, where it may be captured by liver cells and once again secreted into the bile.
Bile acids are taken up by active transport in the terminal ileum, that is, in the lowermost section of the small intestine. The efficiency of reuptake is normally > 90 %. Only the fraction that is not recovered needs to be replaced by de novo synthesis from cholesterol.
During their repeated passages through the intestine, some bile acids undergo modification by microbial enzymes; an example is the formation of deoxycholate from cholate. Such modified molecules become part of the circulating bile acid pool.
| 11.5.3 |
Bile acid cycling involves multiple transport proteins |
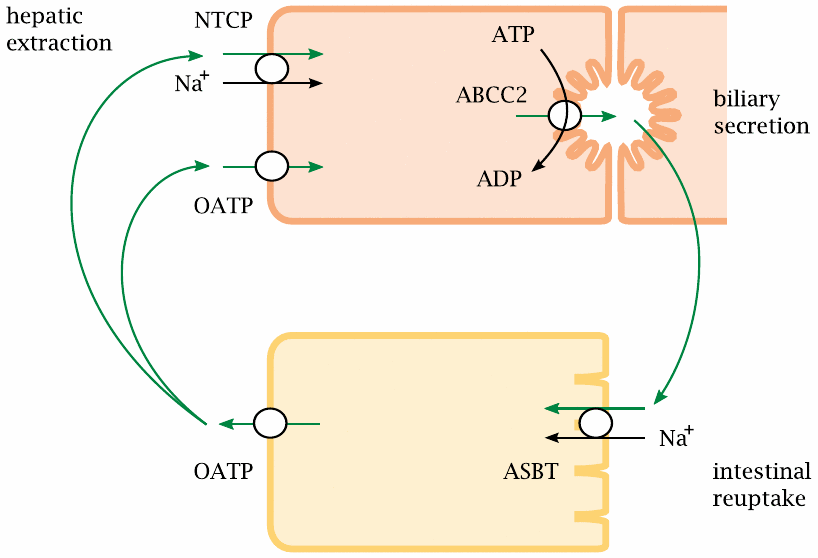
A variety of transport proteins enable the bile acid enterohepatic cycle. Secretion from the liver cell into the bile is driven by ABCC2, another ABC type transporter (compare slide 11.4.5). Reuptake from the lumen of the gut is mediated by the apical sodium-coupled bile acid transporter (ASBT). A similar transporter, the Na+-dependent taurocholate cotransporting polypeptide (NTCP), mediates uptake from the blood back into the liver cell. At the basolateral membranes of both intestinal and liver cells, organic anion transport proteins (OATPs), which have a fairly low degree of substrate specificity, participate in bile acid transport.
| 11.5.4 |
A deficient ABCC2 transporter causes Dubin-Johnson syndrome |
- impaired excretion of bile acids → cholesterol precipitates in the bile → bile stones
- impaired excretion of bilirubin → jaundice
- impaired excretion of many drugs → potential drug toxicity
Compared to other hereditary gene defects, this one is relatively frequent. As noted above, the liver can secrete some surplus cholesterol into the bile, where it is kept in solution by bile acids. A shortage of bile acids promotes its precipitation, which ultimately leads to gallstones. However, cholesterol precipitates also tend to form without the transporter defect, and the sterol is indeed the most common gallstone ingredient.
Apart from bile acids, ABCC2 also secretes the conjugated form of bilirubin (bilirubin diglucuronide, see slide 17.4) as well as the conjugated forms of several drugs (section 19.3). In Dubin-Johnson patients, the retention of bilirubin leads to jaundice, whereas the reduced rate of drug elimination increases their concentration in the system and can lead to toxicity. Such drug toxicity may be prevented by reducing the dosage, or by using drugs that are mostly eliminated through the kidneys. Overall, with proper management, Dubin-Johnson syndrome is not very severe.
| 11.6 |
Cholesterol and atherosclerosis |
| 11.6.1 |
Is atherosclerosis a metabolic disease? |
This quote sums it up rather well—cholesterol metabolism is a key element in the pathogenesis of atherosclerosis. Another key factor is blood pressure, whose role is illustrated by the simple observation that atherosclerosis afflicts the arteries but not the veins. The accumulation of lymphocytes and macrophages in atherosclerotic lesions highlights the role of inflammation.
In keeping with the scope of these notes, we will here focus on the metabolic causes of atherosclerosis and consider any other factors only in broad outline.
| 11.6.2 |
Macroscopic appearance of atherosclerotic lesions |
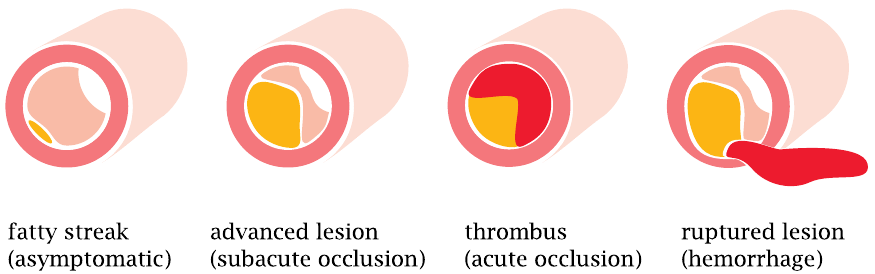
The earliest readily visible atherosclerotic lesion is the fatty streak. It forms within the wall of an artery, in the thin layer of connective tissue that is located underneath the endothelium (the innermost cell layer of any blood vessel) and atop the thick layer of smooth muscle that maintains the wall tension and blood pressure.
Fatty streaks are very common—they will be found in the arteries of virtually any middle-aged to elderly person. As such, a fatty streak does not constitute a problem. However, it tends to progress to more serious stages with time. These advanced lesions wreak havoc in various ways:
- 1.Once they become large, they will constrict the lumen of the artery and hence reduce the blood flow.
- 2.The endothelium atop an advanced lesion may erode. An intact endothelium inhibits blood clot formation; endothelial lesions bring the blood into contact with tissues and molecules that promote blood coagulation. A blood clot, or thrombus, that forms atop such an eroded lesion will cause acute occlusion of the artery. This is what happens in myocardial infarction and in most cases of stroke.
- 3.An advanced lesion may damage not only the endothelium but also the muscular layer of the arterial wall, which may then rupture. Approximately 20% of all cases of stroke arise in this manner.
Atherosclerosis is a systemic disease. While most commonly manifest in the heart and the brain, vascular occlusion and infarction can strike anywhere and everywhere; for example, constriction of arteries in the legs causes leg muscle pain even under light exercise (walking), which is known as intermittent claudication.72
| 11.6.3 |
Microscopic appearance of atherosclerotic lesions |
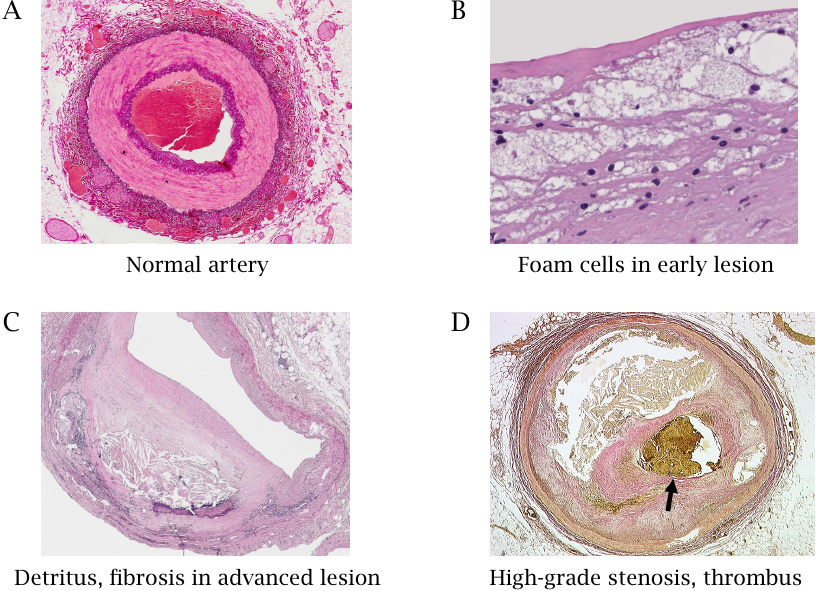
This slide shows cross sections of a normal artery (A) and of atherosclerotic lesions in different stages of advancement (B–D). The normal artery displays inner and outer layers of connective tissue, stained in dark purple, as well as a strong intermediate muscular layer that shows up in a lighter shade. The endothelium is too thin to be discerned at this low power of magnification. The blood clot in the lumen is a post-mortem artifact.
Panel B shows a higher power view of an early lesion. The bubbly appearance is due to foam cells, which are macrophages stuffed chock-full with lipids.73 Panel C shows an advanced lesion with connective tissue proliferation and accumulation of detritus within the vessel wall; the lumen of the artery is considerably constricted. Panel D shows an artery that was already almost completely obliterated by a proliferating lesion that had encompassed the entire circumference; the narrow residual lumen is blocked by an acutely formed thrombus (stained yellow-brown).
| 11.6.4 |
Development of an atherosclerotic lesion |
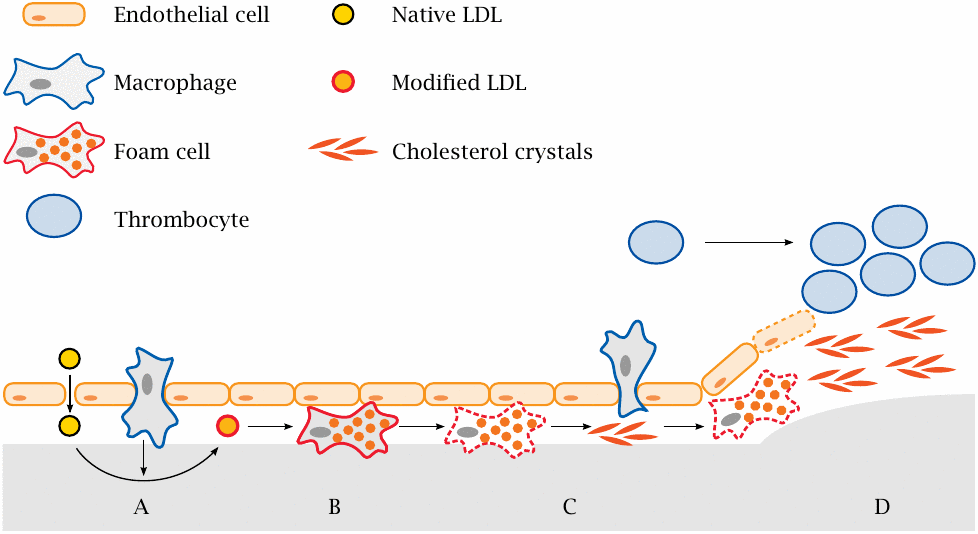
This slide summarizes the developmental stages of an atherosclerotic lesion. A key role is played by low density lipoprotein (LDL), a specific type of plasma lipoprotein particles that is rich in cholesterol (see slide 11.4.2).
- (A)High blood pressure promotes the formation of small defects in the endothelium. These initial lesions allow blood plasma carrying LDL to seep into the subendothelial tissue; this is followed by transmigration of macrophages. Reactive oxygen species and enzymes released by macrophages modify the LDL.
- (B)Modified LDL is taken up by macrophages. Lipid overload turns macrophages into foam cells.
- (C)Foam cells perish, disintegrate and release the accumulated cholesterol, which forms crystalline deposits. These crystals activate new macrophages and cause them to release inflammatory cytokines that further incite and amplify the inflammation [71].
- (D)In an advanced lesion, cells in the muscular layer proliferate, progressively constricting the artery. When the endothelium that covers the lesion becomes eroded, thrombocytes and plasmatic coagulation factors are activated and initiate blood clotting, causing acute thrombus formation and obstruction.
As an alternative to thrombus formation, acute failure of a damaged artery can also occur through rupture. As stated above, approximately 20% of all cerebral infarctions are caused by rupture and hemorrhage rather than thrombotic occlusion. Hemorrhage is less common in other organs.
The proinflammatory activity of cholesterol crystals echoes that of other crystalline deposits with different chemical composition, such as urate crystals in gout [72] and silica crystals in silicosis (miner’s lung). Intracellular signaling complexes called inflammasomes are involved in the reaction in each case.
| 11.6.5 |
Metabolic aspects of atherosclerosis |
- cholesterol uptake, synthesis and degradation
- cholesterol transport in the circulation: LDL (low density lipoprotein) and HDL (high density lipoprotein)
- biochemical changes that turn physiological, benign LDL into an atherogenic agent
| 11.6.6 |
Two modes of uptake of cholesterol into macrophages |
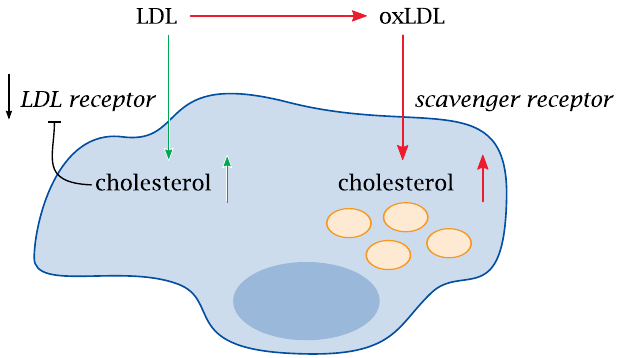
With macrophages as with other cell types, uptake of native LDL via the LDL receptor is regulated by negative feedback, which lets the cells avoid cholesterol overload. The downregulation of the LDL receptor is achieved by transcriptional regulation via the SREBP/SCAP pathway (section 11.3).
In contrast to other cells, however, macrophages also have so-called scavenger receptors, through which they bind and ingest various kinds of debris. Scavenger receptors do not take up native LDL and are not subject to cholesterol-dependent regulation. Oxidized or otherwise modified LDL, however, does enter via the scavenger receptors, which induces cholesterol overload and transforms the macrophages to foam cells. This is a crucial step in the pathogenesis of atherosclerosis.
| 11.6.7 |
Experimental protein modifications that turn LDL into a ligand for the scavenger receptor |
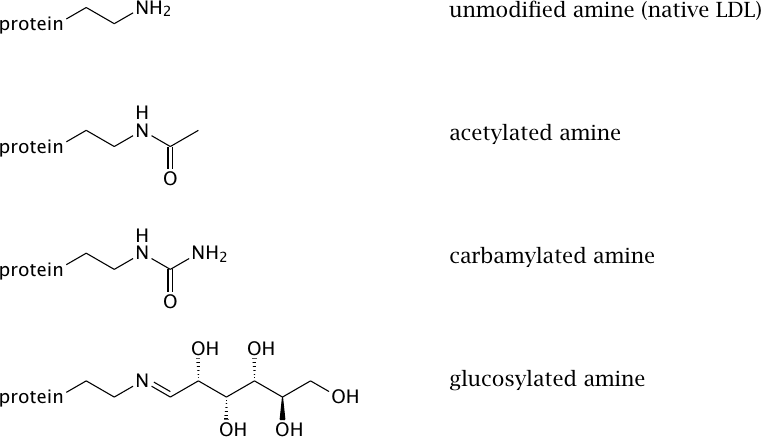
In order to better understand what processes might turn LDL into a scavenger receptor ligand, various types of chemical modifications have been applied in vitro. All of the experimental modifications shown here affect the amino groups of the protein component of LDL (apolipoprotein B). They do not mimic oxidation; therefore, these findings show that LDL oxidation is not the only mechanism that may cause pathologically increased uptake of cholesterol into macrophages. Some, but not all of these modifications are likely to occur in vivo.
| 11.6.8 |
Which modifications of LDL are significant in vivo? |
| Modification | Possible causes |
| acetylation | easily achieved in vitro, but not plausible in vivo |
| carbamylation | promoted by urea, which is enhanced in kidney disease; also promoted by smoking |
| glucosylation | promoted by high blood glucose (diabetes) |
| partial proteolysis | proteases released from macrophages |
| oxidation of lipids and apolipoproteins | reactive oxygen species released from macrophages |
Oxidized LDL has been demonstrated inside atherosclerotic lesions. Oxidation is widely considered to be the most important single mechanism of LDL modification. It is, however, not very well characterized in molecular terms, and the relative importance of protein and lipid oxidation is not clear.
Carbamylation is due to the reaction of amino groups with isocyanate (HN=C=O), which arises through isomerization from urea. Isocyanate apparently also forms from isothiocyanate (HN=C=S) through oxidation, and it has been proposed that this mechanism promotes atherosclerosis in smokers [73].
Glucosylation is likely increased in diabetes and may contribute to the observed acceleration of atherosclerosis in diabetic patients, although it will be difficult to separate this effect from that of other metabolic consequences of diabetes such as hyperlipidemia. The mechanism of glucosylation is the same as with that of hemoglobin (see section 14.5.8).
Proteases are released by inflammatory cells, which occur in atherosclerotic lesions; therefore, partial proteolytic degradation is another mechanism that is plausible in vivo[74].
| 11.6.9 |
How does LDL become oxidized? |
- Phagocytes produce reactive oxygen species
- Transition metals (Fe, Cu) exacerbate ROS activity
- Lipoxygenases convert fatty acids to radicals that can bind to LDL and induce lipid peroxidation
Macrophages and granulocytes produce reactive oxygen species, and iron and copper ions may convert relatively benign ROS (O2•−, H2O2) to more aggressive ones (•OH); the mechanisms are explained in section 18.3. Phagocytes also contain myeloperoxidase, which reacts H2O2 with chloride ions to generate bleach (HOCl). Bleach can react with tyrosine residues, and chlorotyrosine has indeed been detected in oxidized LDL samples obtained from humans. Animal experiments suggest, however, that this mechanism does not significantly promote the development of atherosclerotic lesions (see below).
Lipoxygenases attach molecular oxygen to arachidonic acid and similar polyunsaturated fatty acids (see slide 18.5.8). The hydroperoxy-radicals thus generated can then associate with LDL and set off a self-sustaining cycle of lipid peroxidation, which requires nothing more than a supply of molecular oxygen to turn large numbers of unsaturated fatty acids into their hydroperoxide derivatives. This cycle is discussed in section 18.5; its control by vitamin E and other lipophilic antioxidants is explained in slide 18.7.11f.
| 11.6.10 |
Experimental evidence implicating LDL oxidation in the pathogenesis of atherosclerosis |
- Vitamin E reduces the severity of atherosclerosis in animal models—but not in clinical studies on humans
- Antibodies against oxidized LDL are found in blood; among these, IgG promotes atherosclerosis, whereas IgM inhibits it
- Haptoglobin alleles differ in the efficiency of hemoglobin clearance, which correlates inversely with susceptibility to atherosclerosis
- Production of HOCl by myeloperoxidase: chlorotyrosine residues detectable in oxLDL ex vivo—but myeloperoxidase k.o. mice have increased susceptibility to atherosclerosis
Regarding vitamin E: It never ceases to amaze me how many therapies that work wonders in mice fall flat when applied to humans. Too bad we aren’t mice … maybe after all Douglas Adams [75] was right about who was running the show between man and mice?74
The effect of antibodies to oxidized LDL may be due to opsonization: antibody-decorated particles are taken up more efficiently by phagocytes, which have receptors for antibodies on their surface. Since there are receptors for both IgG and IgM, I’m not sure what to make of the observed difference between the two types of antibodies on atherosclerosis progression. In any case, the observation that antibodies have any effect at all suggests that their antigen matters.
Haptoglobin is a serum protein that captures hemoglobin released from decayed erythrocytes. In adults, the regular turnover of red blood cells releases several grams of hemoglobin every day. If free hemoglobin is not promptly cleared, it may shed heme, which then may bind to LDL and promote its oxidation [76]. Haptoglobin genotypes are associated with the risk to suffer from complications of atherosclerosis [77].
Myeloperoxidase produces HOCl from H2O2. While oxidation of LDL by HOCl may promote LDL phagocytosis, the effect of genetic myeloperoxidase knockout suggests that HOCl is less effective in this regard than the H2O2 consumed in its formation would have been.
| 11.7 |
Cholesterol metabolism and the treatment of atherosclerosis |
| 11.7.1 |
Lowering LDL cholesterol: therapeutic principles |
- inhibition of cholesterol synthesis
- inhibition of cholesterol uptake
- inhibition of cholesterol ester transfer protein
- inhibition of bile acid reuptake
- LDL apheresis
Lowering cholesterol is a key objective in the clinical management of atherosclerosis. Based on the foregoing, we can appreciate the strategies listed here. Two or more of these strategies are often used in combination.
| 11.7.2 |
“Statins” inhibit HMG-CoA reductase |
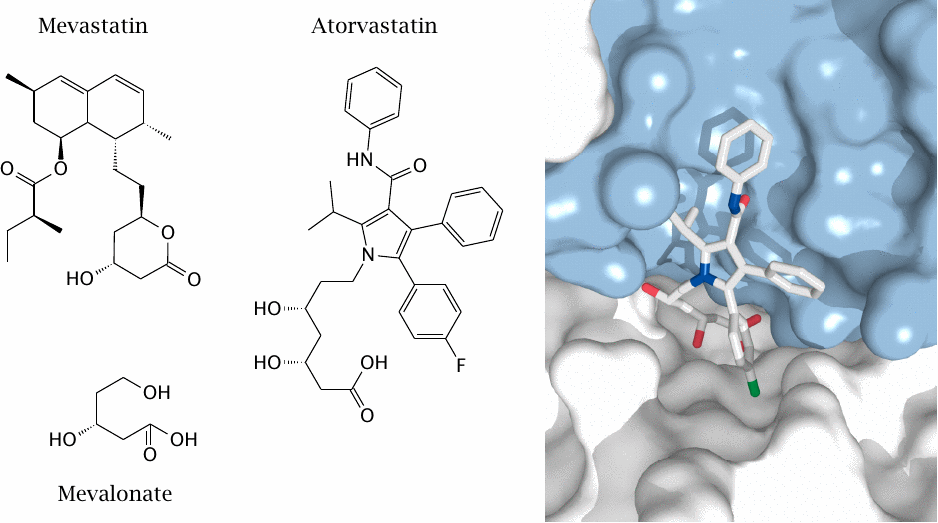
Inhibition of HMG-CoA reductase is currently the single most effective and widely used strategy to lower plasma cholesterol. The first inhibitor was mevastatin, a natural compound isolated from the fungus Penicillium citrinum. Its function in nature is probably to inhibit the growth of competing fungal species by blocking their synthesis of ergosterol, which also requires HMG-CoA reductase.
Some early therapeutically useful statins, such as lovastatin, were derived from mevastatin. Modern statins such as atorvastatin depart from the mevastatin structure. However, both mevastatin and atorvastatin contain a moiety resembling the product of the enzyme reaction (mevalonate).
HMG-CoA reductase has four subunits, with four active sites located at subunit interfaces. The picture shows one active site confined between two subunits, which are rendered in white and blue, respectively. An atorvastatin molecule occupies the active site. Rendered from 1hwk.pdb.
| 11.7.3 |
Inhibitors of intestinal cholesterol uptake |
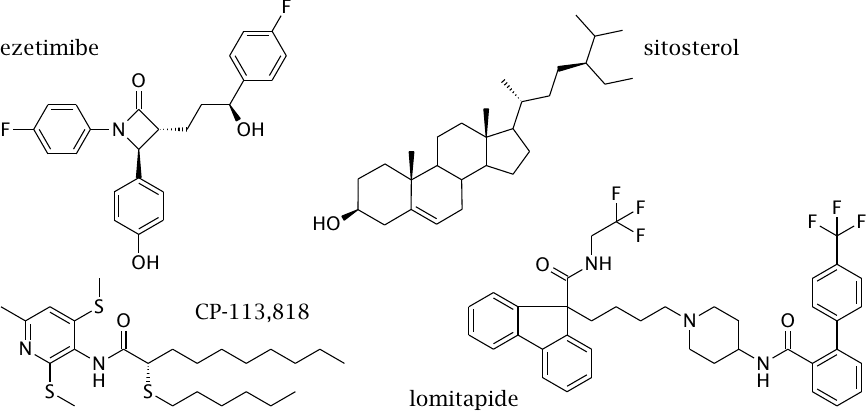
Inhibitors of intestinal cholesterol absorption represent another plausible strategy to lower plasma cholesterol, but their effect on blood cholesterol levels is less powerful than that of the statins.
Ezetimibe binds to the NPC1L1 protein in the luminal cell membrane (slide 11.4.3) and inactivates it. The drug is used mostly in conjunction with statin drugs. The β-lactam ring in the center of the structure is likely reactive, which suggests that ezetimibe may bind its target covalently, but I have not found experimental evidence supporting this assumption.
Lomitapide inhibits the transfer of cholesterol from endocytotic vesicles to the ER by mitochondrial triglyceride transfer protein (MTTP). At the ER, cholesterol is normally converted to cholesterol esters by ACAT. CP-113,818 is an experimental inhibitor of ACAT. These two strategies are currently still experimental.
Sitosterol and other plant sterols compete with cholesterol for intestinal uptake. This “low tech” strategy is clinically proven yet has modest benefits.
| 11.7.4 |
Cholesterol ester transfer protein (CETP) short-circuits cholesterol transport by lipoproteins |
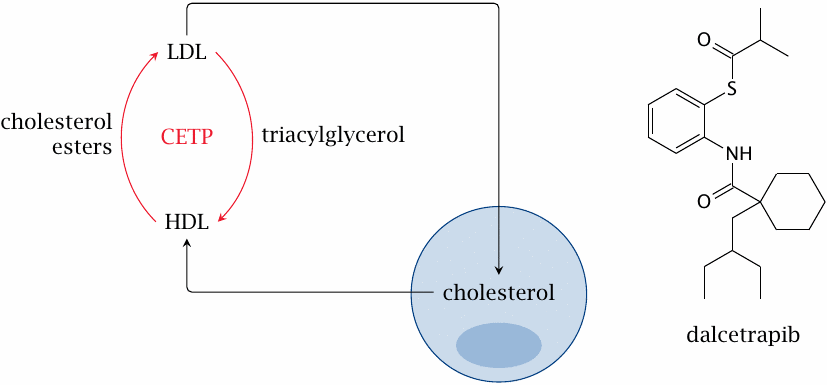
CETP, a serum protein, facilitates the exchange of triacylglycerol and cholesterol esters between HDL and LDL. The net effect seems to be an increase of LDL cholesterol, and accordingly inhibition of CETP looks like a promising strategy.
Dalcetrapib is one of several CETP inhibitors that have been or currently are under study by various pharmaceutical companies. While dalcetrapib indeed raises the ratio of HDL cholesterol to LDL cholesterol, large scale clinical trials have been put on hold due to lack of improvement in clinical outcomes [78].
| 11.7.5 |
Cholestyramine particles absorb bile acids |
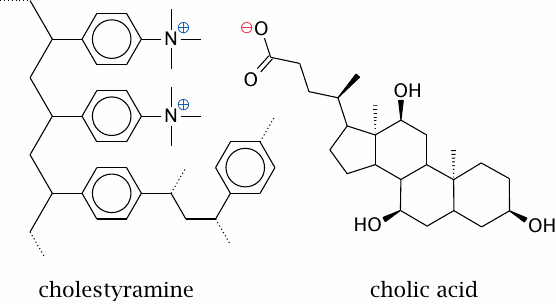
Cholestyramine and similar polymers adsorb bile acids due to a combination of electrostatic and hydrophobic forces. This prevents the bile acids from being taken up at the end of the small intestine. They are replaced by de novo synthesis from cholesterol, which therefore depletes the pool of cholesterol in the liver.
The strategy is effective but has some side effects. The concentration of bile acids in the bile is reduced; this promotes precipitation of cholesterol and other poorly soluble bile constituents, which may then form gallstones. The polymer particles may also bind some drugs or fat-soluble vitamins and prevent their absorption.
| 11.7.6 |
LDL apheresis |
- Blood is diverted through an extra-corporeal filtration device
- cells are separated from plasma
- LDL is removed from plasma by affinity methods or size-based filtration
- The remaining plasma and cells are returned to the circulation
- The procedure is repeated in weekly or biweekly intervals
While conceptually simple, LDL apheresis is involved and time-consuming in practice. It is therefore used only in severe cases, such as homozygous familial hypercholesterolemia.
| 11.7.7 |
More … |
- triparanol—an old drug, inhibits some CYP450 enzymes in the conversion from lanosterol to cholesterol; withdrawn due to toxicity
- bezafibrate—a PPARγ agonist
- nicotinic acid—activates hormone-sensitive lipase through a G protein coupled receptor named HM74A; 5 likely additional mechanisms
- probucol and succinobucol—supposedly antioxidants that prevent LDL oxidation, but also cause unrelated changes in other laboratory parameters
- guar gum and other carbohydrate fibers —absorb and prevent intestinal uptake of cholesterol and bile acids with variable efficiency
- thyroid hormone analogs—promote LDL utilization
We will not cover these drugs in detail, but you may find it interesting to look them up in the literature yourself.
| 11.8 |
Gene defects in cholesterol transport and metabolism |
| 11.8.1 |
Familial hypercholesterolemia is due to a gene defect in the LDL receptor |
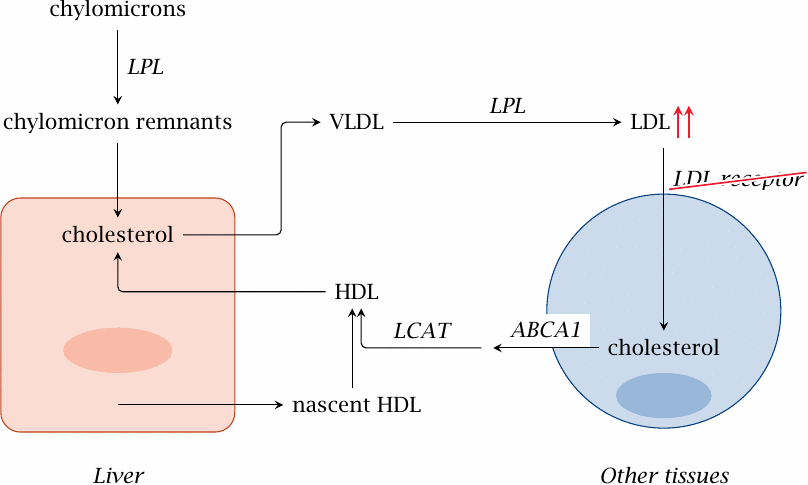
In Caucasians, this gene defect occurs with a heterozygote frequency of 1 in 500. While most other hereditary defects of metabolism are clinically manifest only in homozygous form, familial hypercholesterolemia is symptomatic in both heterozygous and homozygous individuals, and therefore much more commonly encountered in clinical practice.
The defect is more severe in homozygous patients, whose levels of LDL in the blood are several times higher than normal. Before effective treatment options became available, these patients used to develop severe atherosclerosis at young age, leading to death due to myocardial infarction or stroke by the age of 40 years or earlier. This situation has improved through therapy with statin drugs and LDL apheresis.
| 11.8.2 |
Tangier disease: Disruption of cholesterol transfer to HDL |
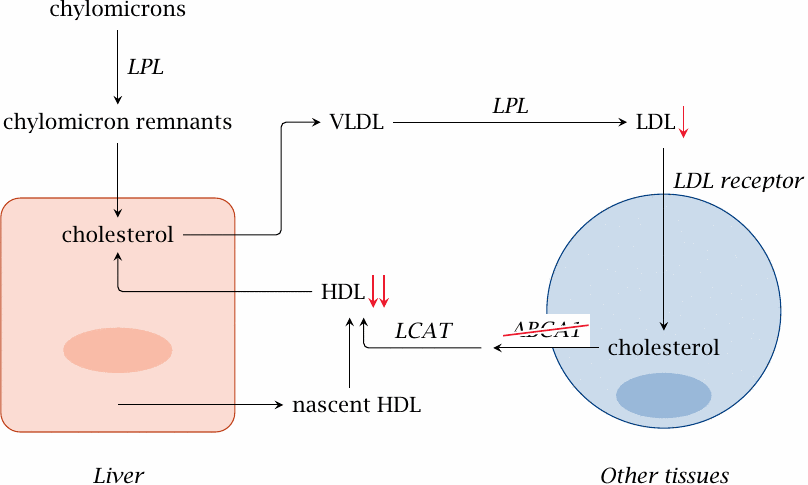
Tangier disease, which is very rare, is interesting from a mechanistic point of view. The defect concerns the ABC transporter that exports surplus cholesterol from the cell for delivery to HDL. As a consequence, HDL is greatly reduced. Interestingly, LDL cholesterol is also reduced; this may be due to the transfer of cholesterol between LDL and HDL mediated by CETP (see slide 11.7.4).
Fatty deposits are found in the liver, spleen and cornea. Patients develop atherosclerosis, type 2 diabetes, and neuropathy.
| 11.8.3 |
A defective plant sterol exporter causes sitosterolemia |
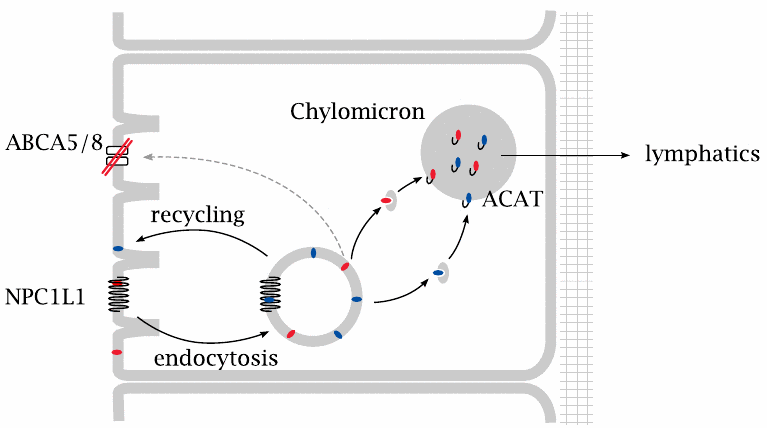
Genetic deficiency of the ABCA5/8 transporter results in sitosterolemia. Plant sterols that are taken up from the gut lumen can no longer be extruded, accumulate inside the mucosal cells, and ultimately find their way into the chylomicrons and the system. Patients have very high levels of plant sterols in the plasma; they develop lipid deposits in the skin and atherosclerosis.
In these patients, therapeutic application of plant sterols in order to inhibit cholesterol uptake would obviously be a bad idea. The treatment of choice is ezetimibe [79,80], which inhibits the NPC1L1 protein (see slide 11.7.3).