| 13 |
Hormonal regulation of metabolism |
| 13.1 |
Hormones that affect energy metabolism |
| Hormone | Message |
| insulin | glucose and amino acids available, more substrates on the way |
| glucagon | glucose and amino acids in short supply, need to mobilize internal reserves |
| epinephrine | prepare for imminent sharp rise in substrate demand |
| glucocorticoids | prepare for extended period of high demand |
| thyroid hormones | increase basal metabolic rate |
We have already touched briefly on the effects of insulin, glucagon and epinephrine on gluconeogenesis and glycogen metabolism. In this chapter, we will take a more thorough look at these hormones’ properties and activities. This will provide the foundation for the next chapter, in which we will consider the disruptions of metabolic regulation that occur in diabetes mellitus.
Of the hormones listed in the table, only insulin has the effect of lowering blood glucose. The other hormones are all antagonistic to insulin, and a pathological increase in their secretion may result in symptomatic diabetes. In the case of glucocorticoids, symptomatic diabetes may also arise from their use as drugs.
Both glucocorticoids and thyroid hormones cause their effects mostly through up- and down-regulating gene transcription. The two types of hormones control different sets of target genes, but both upregulate the expression of β-adrenergic receptors, that is, receptors for epinephrine, and so amplify the metabolic effects of this hormone.
| 13.2 |
Insulin |
| 13.2.1 |
Langerhans’ islets in the pancreas produce insulin and glucagon |
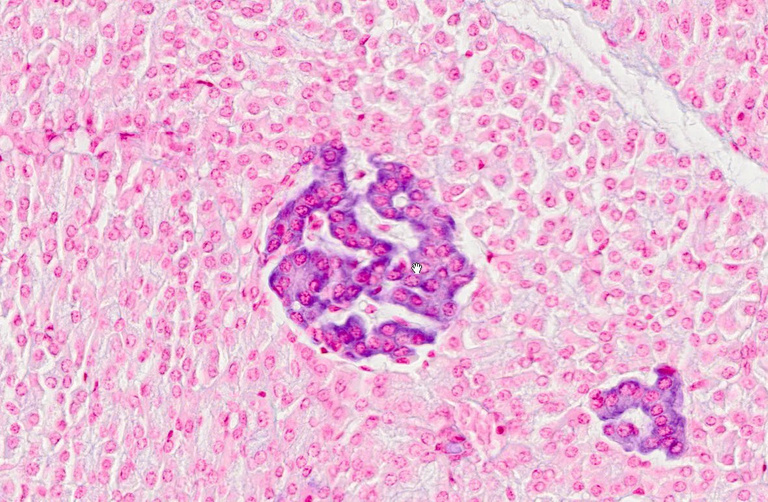
Insulin is produced in the islets of Langerhans, which are small clusters of cells that occur interspersed within the exocrine pancreas. In this picture, one islet is shown in the center, and a tangential section across another is seen in the bottom right. The two islets are embedded in a “red sea” of exocrine pancreas tissue.
The function of the exocrine pancreas—secretion of digestive enzymes and of bicarbonate into the small intestine—has been discussed in slide 1.6.7. The products of islet cells are secreted into the bloodstream; therefore, the islets collectively function as an endocrine gland. Among the several cell types found in the islets, the β-cells produce insulin, whereas the α-cells produce glucagon.
| 13.2.2 |
A little bit of history: The purification of insulin—the problem |
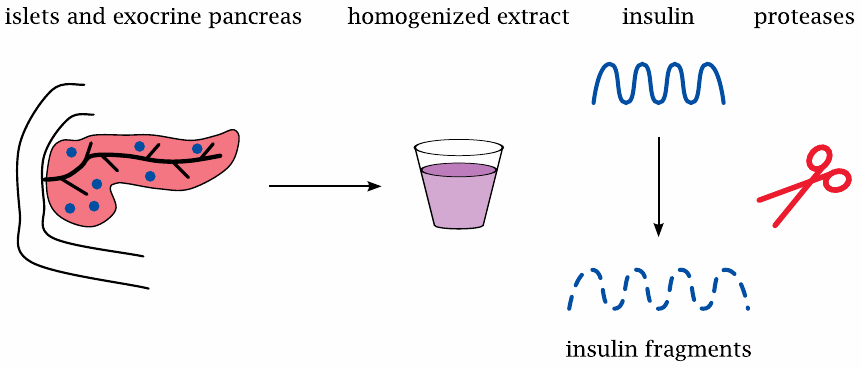
When purifying a protein of interest from some organ, standard practice is to first grind up the tissue with a homogenizer, and then to subject the homogenate to various fractionation procedures so as to incrementally enrich the protein, until it is sufficiently clean. With pancreatic tissue, homogenization releases large amounts of proteases from the exocrine gland cells; these enzymes will then chop up any other protein contained in the homogenate before it can be purified. Once it had been recognized that pancreatic islets must contain an anti-diabetic hormone, the proteases present in the pancreas homogenates foiled initial attempts to purify it.85
| 13.2.3 |
The purification of insulin—Banting’s solution |

The bright idea that overcame this problem occurred, in one of his sleepless nights, to a young physician named Frederick Banting. He immediately resolved to pursue this idea at the University of Toronto, where he was joined in this effort by a junior colleague, Charles Best.
It had previously been observed in human patients that occlusion of the pancreatic duct induced a protracted self-destruction of the exocrine pancreas tissue, while sparing the pancreatic islets. Banting surgically obstructed the pancreatic ducts of experimental animals so as to induce destruction of the exocrine gland tissue. The source of the contaminating proteases was thus removed, and Banting and his colleagues were able to extract and purify insulin from the islets that remained intact within the biochemically inert scar tissue that had replaced the exocrine pancreas.
It is worth noting that Banting and his colleagues did not proceed to claim a patent, but instead shared their discovery freely, encouraging everyone else to use it. This generous act ensured that the diabetics of the world were soon supplied with insulin.
| 13.2.4 |
Historical side note: Norman Bethune, Banting’s famous classmate |

Of historical interest, but not relevant to the main subject, is that one of Banting’s classmates at medical school was Norman Bethune, a gifted surgeon who invented many surgical instruments and introduced mobile blood transfusion units in the Spanish civil war. He subsequently joined an armed unit of the Chinese Communist Party that fought in the Sino-Japanese war.
The picture on the left shows Bethune performing surgery in a makeshift hospital in the Chinese countryside. During one such operation, Bethune contracted a bacterial infection to which he subsequently succumbed. The picture on the right shows Bethune’s birthplace in the city of Gravenhurst, which is located in Ontario’s Muskoka region. It is now a museum and is well worth a visit.
| 13.2.5 |
Structure of insulin and its precursors (1) |
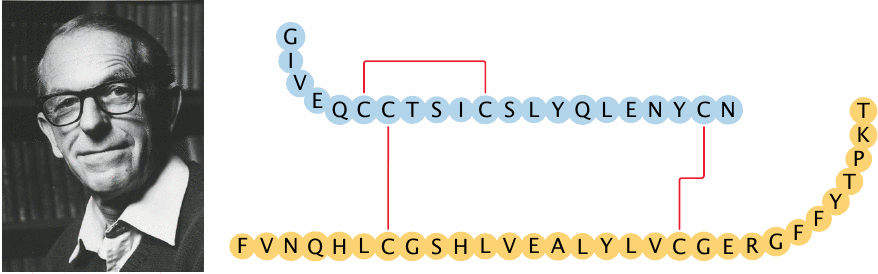
The insulin molecule consists of two peptide chains, which are held together by two disulfide bridges. The sequence of insulin was the very first protein sequence ever to be determined, and the methodology for this feat was developed by the gentleman with the sardonic smile, Frederick Sanger.
Sanger went on to develop similarly ingenious methods for sequencing RNA and DNA. His own account of these discoveries [85], appropriately entitled “Sequences, sequences and sequences,” is quite humorous and well worth reading.
| 13.2.6 |
Structure of insulin and its precursors (2) |
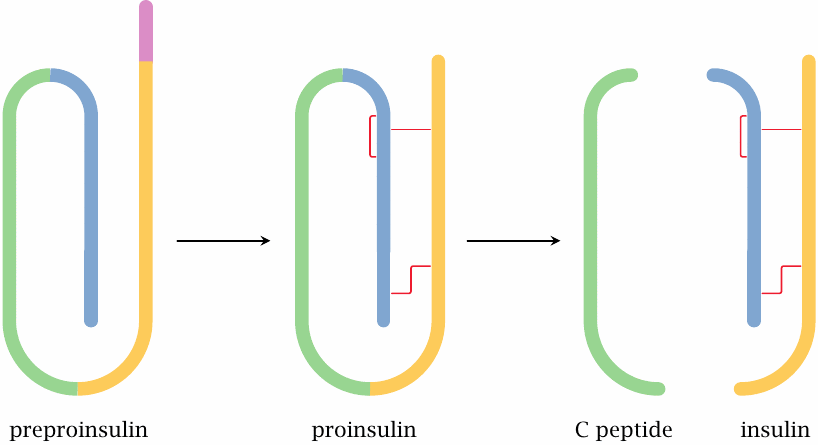
Pre-proinsulin is the primary translation product, as it runs off the ribosome at the membrane of the rough endoplasmic reticulum. It still contains the N-terminal signal peptide and lacks the disulfide bridges. The signal peptide is cleaved off, and the disulfide bonds are formed en route from the ER via the Golgi apparatus to the secretory vesicles. The C-peptide is cleaved off as well, but it remains inside the vesicles and is secreted together with insulin.
C-peptide has for a long time been considered physiologically irrelevant; it was used only as a diagnostic parameter to assess the residual secretory function of β-cells in type 2 diabetics. However, several recent experimental studies support the idea that C-peptide itself is active as a growth factor, and that its lack in type 1 diabetes contributes to the development of diabetic long-term complications. On cell surfaces, saturable binding sites have been characterized, suggesting the existence of a specific receptor; however, no receptor has yet been purified. In small-scale pilot studies on type 1 diabetics, beneficial effects have been observed when insulin was supplemented with C-peptide. However, these studies have not yet translated into routine therapy of diabetes [86].
| 13.2.7 |
Sequences of human, swine, and bovine insulins |
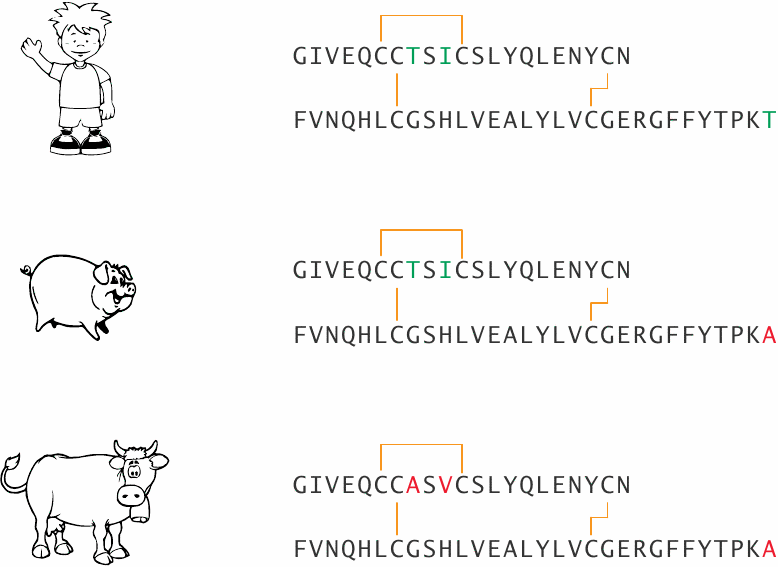
Until the late 1980s, swine and cow insulins were the mainstay of insulin substitution therapy. They are fully active in humans but differ from human insulin in one or three amino acid positions, respectively. This difference may promote the formation of antibodies, which bind and inactivate insulin. Recombinantly expressed human insulin has replaced the animal insulins in therapy, which has largely done away with this problem.86
| 13.2.8 |
Insulin secretion in the β-cell is controlled by glucose and triggered by membrane depolarization |
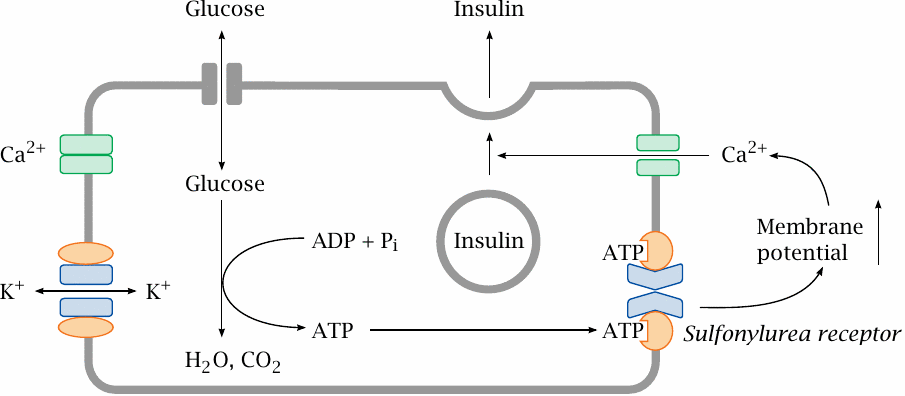
While insulin controls the rate of glucose uptake in many tissues, the β-cells, which control the release of insulin, themselves take up glucose in an insulin-independent manner; the rate of uptake cells thus simply depends on the plasma glucose level. Inside the β-cells, glucose undergoes degradation, which increases the cellular level of ATP. The ATP then binds to the sulfonylurea receptor, which in turn closes a potassium channel associated with it. This leads to an increase in the membrane potential, which activates a voltage-gated Ca++ channel. Ca++ entering the cell triggers the exocytosis of insulin and C-peptide.
Glucose is not the only substrate that can be degraded to yield ATP, and it is not surprising that some amino acids and fatty acids will also promote insulin secretion. In addition, several types of cell surface receptors contribute to the activation of insulin secretion in a manner that is not dependent on substrate degradation.
| 13.2.9 |
The sulfonylurea receptor controls an associated potassium channel |
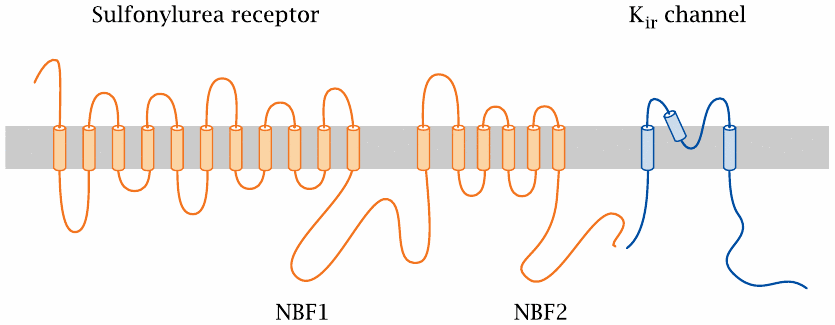
This figure illustrates how the polypeptide chains of the sulfonylurea receptor and of the associated “inward rectifier” Kir channel crisscross the cell membrane. The N-terminus of the sulfonylurea receptor is extracellularly located. NBF1 and NBF2 are nucleotide-binding folds, that is, conserved protein sequence motifs that are involved in the binding of ATP.
The scheme shows a single Kir molecule; however, a functional K+ channel consists of four Kir subunits. The entire ensemble of Kir and sulfonylurea receptor subunits is referred to as a KATP channel.
| 13.2.10 |
KATP channels also regulate the tone of smooth muscle cells |
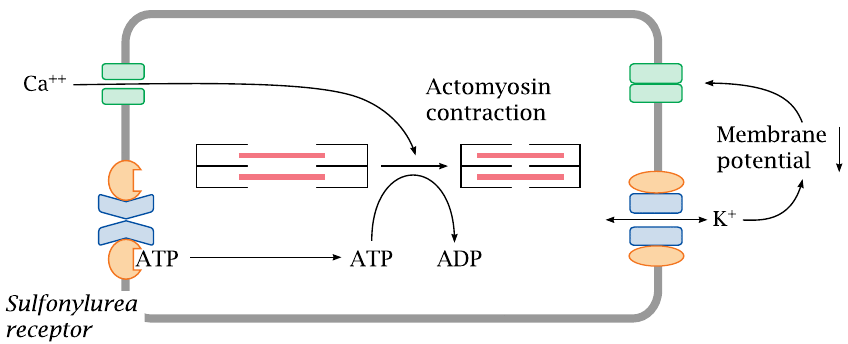
KATP channels serve in more than one physiological role. In vascular smooth muscle cells, KATP channels regulate the strength of contraction. Sustained contraction of a muscle cell will reduce its ATP level, which will promote dissociation of ATP from the sulfonylurea receptor and open the connected Kir channel. The increase in K+ permeability will lower the membrane potential and inhibit the activation of voltage-gated calcium channels; this, in turn, will inhibit cell contraction. This mechanism protects the cell from excessive exertion: when ATP is depleted, the opening of the KATP channels will cause the cell to ignore any further calcium signals and suspend contraction until it has caught its breath and replenished ATP.
Drugs that counteract the effect of ATP on the sulfonylurea receptor will keep the Kir open and promote relaxation of vascular smooth muscle cells. This is an effective means to lower blood pressure.
| 13.2.11 |
Tolbutamide promotes closing of the KATP channel |
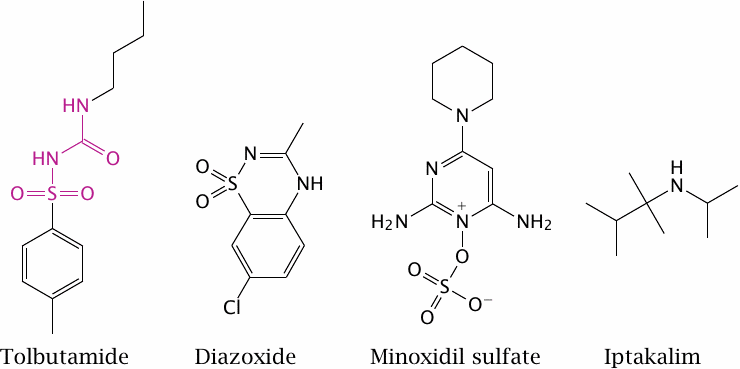
The sulfonylurea receptor is so named because it responds to sulfonylurea derivatives such as tolbutamide. The effect of tolbutamide augments that of ATP, so that a given level of glucose/ATP results in the secretion of more insulin. This is a useful therapeutic principle in type 2 diabetes, as long as the patients’ β-cells remain functional. It is ineffective in type 1 diabetes, since the β-cells are destroyed in this condition (see chapter 14).
The drugs diazoxide and minoxidil contain structurally similar moieties but inhibit the sulfonylurea receptor. They are used to induce vascular relaxation, but diazoxide in particular also reduces insulin secretion as a side effect. The drug iptakalim reportedly activates the vascular KATP channel but inhibits the one on β-cells; this would combine the two beneficial effects. Iptakalim likely interacts directly with the Kir channel rather than with the sulfonylurea receptor.
| 13.2.12 |
The insulin receptor is a receptor tyrosine kinase |
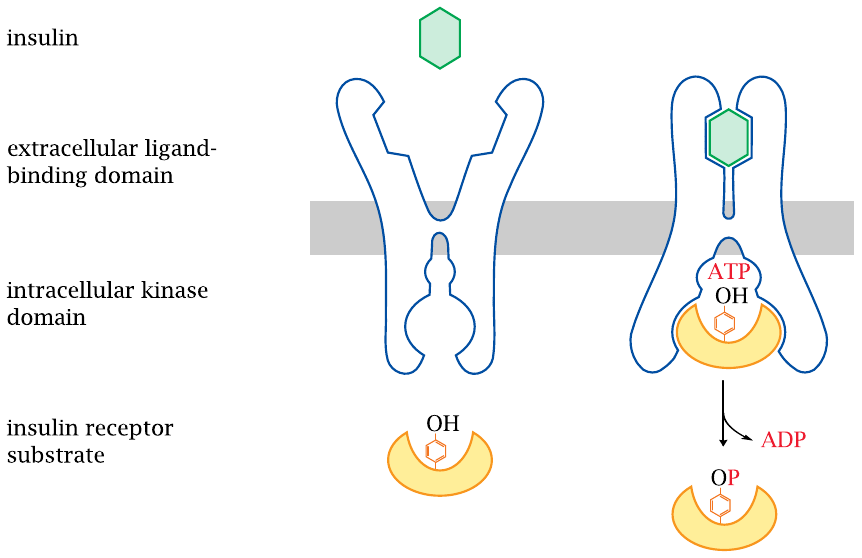
The insulin receptor is found on the surface of all body cells that respond to the hormone. The highest receptor density is found on liver cells; in fact, more than half of the insulin that is released by the pancreas is captured by receptors in the liver (recall that venous blood from the pancreas is drained into the portal vein; see slide 1.6.2).
The insulin receptor is located in the cytoplasmic membrane; it is a receptor tyrosine kinase. Aside from insulin, human growth hormone and many other growth factors have receptors of this type. Receptor tyrosine kinases are one of the major functional classes of hormone receptors.
A receptor tyrosine kinase has two functional domains. The extracellular domain binds to the hormone. This causes a conformational change to the entire receptor, which activates the intracellular protein tyrosine kinase domain. The activated receptor binds one or several cognate protein substrates, which it then phosphorylates at specific tyrosine residues. The phosphorylated substrates leave the receptor and interact with downstream adapter proteins, which then set off various intracellular signaling cascades.
| 13.2.13 |
Insulin receptor first phosphorylates itself and then a number of insulin receptor substrate proteins |
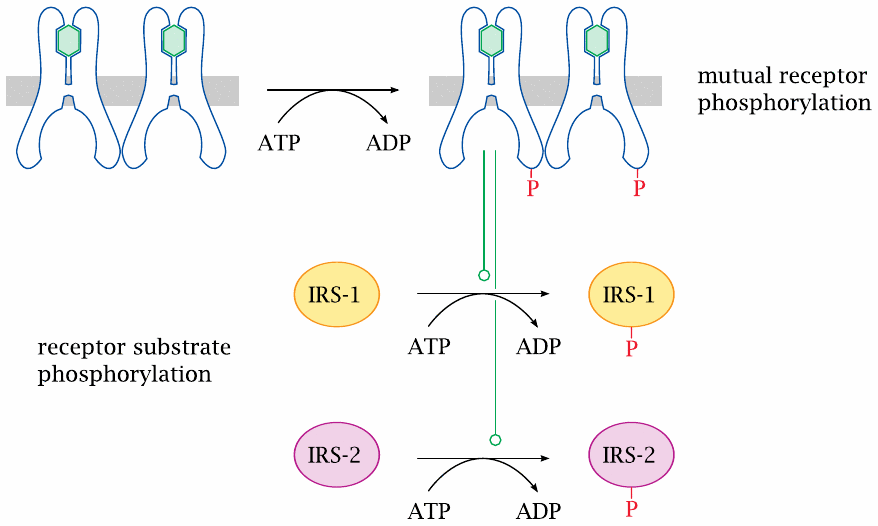
In the case of insulin receptor, the first target for phosphorylation is another insulin receptor molecule; mutual phosphorylation of the two receptors locks both into the active conformation. Subsequently, the receptor molecules phosphorylate a series of regulatory proteins, which are referred to as insulin receptor substrates (IRS).
| 13.2.14 |
Insulin effects on glycogen synthesis |
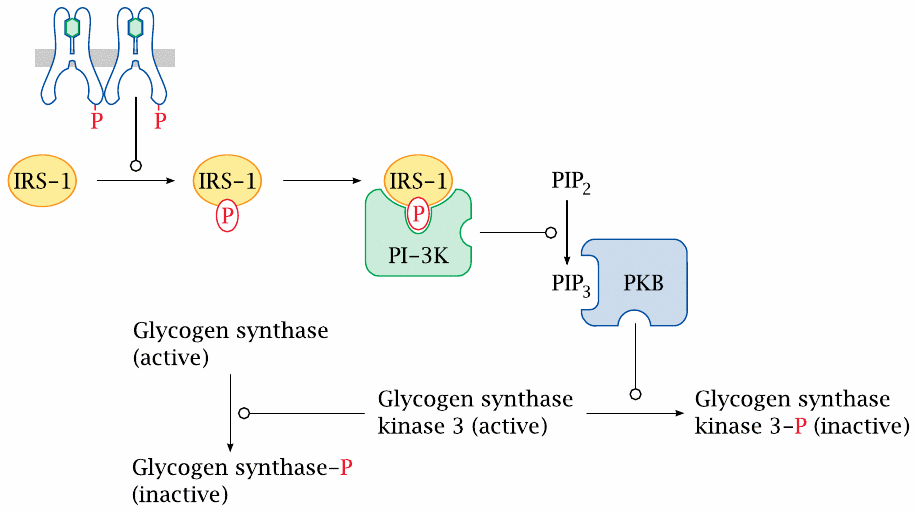
The effect of insulin on glycogen synthesis is mediated through a cascade that involves phosphorylated insulin receptor substrate 1 (IRS-1), phosphatidylinositol-3-kinase (PI-3K), and protein kinase B, which phosphorylates and thereby inactivates glycogen synthase kinase 3. This results in less phosphorylation of glycogen synthase; glycogen synthase itself will therefore be left in the dephosphorylated, active state.
Insulin also activates phosphodiesterase, which lowers cAMP and thereby also affects the phosphorylation of glycogen synthase and of phosphorylase kinase (see slide 8.4.2). This effect is mediated via protein kinase B, too, but the exact molecular cascade from PKB to phosphodiesterase is not clear.
| 13.2.15 |
The role of insulin in glucose transport |
| Active transport | Facilitated transport | |
| insulin-independent | small intestine, kidney tubules | brain, β-cells, red blood cells, cornea and lens of the eye |
| insulin-dependent | never | muscle, fat, most other tissues |
The brain must keep working at all times; it depends on glucose and can take it up from the blood with or without insulin. In contrast, most other tissues can more readily replace glucose with other energy-rich substrates. The uptake of glucose into the cells of those tissues depends on insulin. When glucose supply is low, insulin secretion drops as well. Glucose uptake by insulin-dependent tissues ceases, which preserves glucose for the brain.
If blood glucose drops to excessively low values—this state is called hypoglycemia—the brain will no longer manage to obtain enough glucose, which will lead to unconsciousness and can result in brain damage and death.
| 13.2.16 |
Insulin promotes glucose uptake by increasing the surface exposure of GLUT 4 transporters |
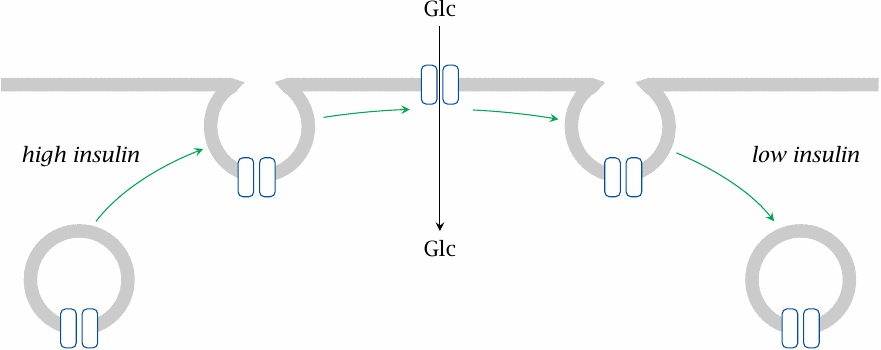
The distinction between insulin-dependent and -independent uptake correlates with different subtypes of the GLUT transporters. The major insulin-dependent type is GLUT 4, whereas GLUT 1 and 2 are insulin-independent. GLUT4 transporters undergo reversible translocation between the cytoplasmic membrane and intracellular storage vesicles. Obviously, only transporters currently residing in the cell membrane can transport glucose. The transporter migration is controlled through insulin-dependent phosphorylation of cytoskeletal proteins downstream of protein kinase B.
Insulin-independent transporters remain in the cell membrane throughout. In diabetes mellitus, cells with insulin-dependent glucose uptake will experience glucose starvation, whereas those with insulin-independent uptake will be exposed to glucose overload.
| 13.2.17 |
Transcriptional regulation by insulin |
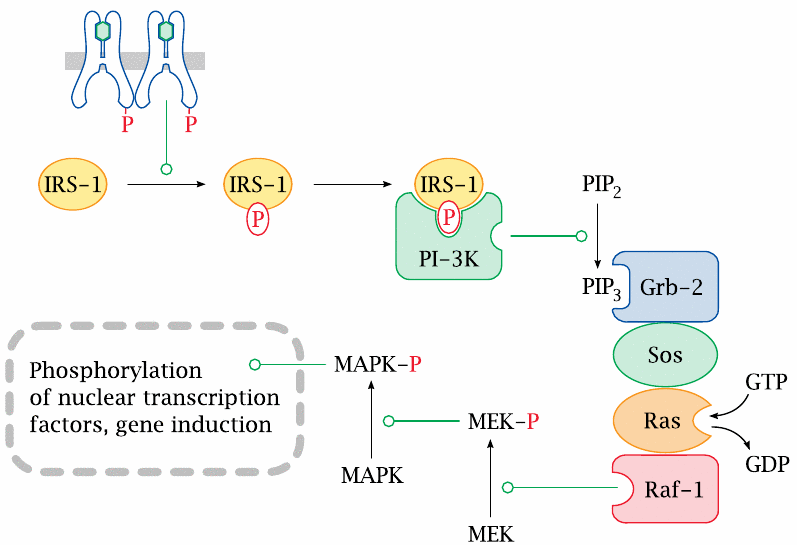
In addition to regulating the activity of pre-formed enzymes and transporters through phosphorylation, insulin also regulates the de novo expression of proteins at the level of transcription.
The participating proteins are numerous and all have suitably intriguing and cryptic names—for example, “Sos” is shorthand for “son of sevenless”—but we won’t go into details about them here.
| 13.3 |
Other hormones |
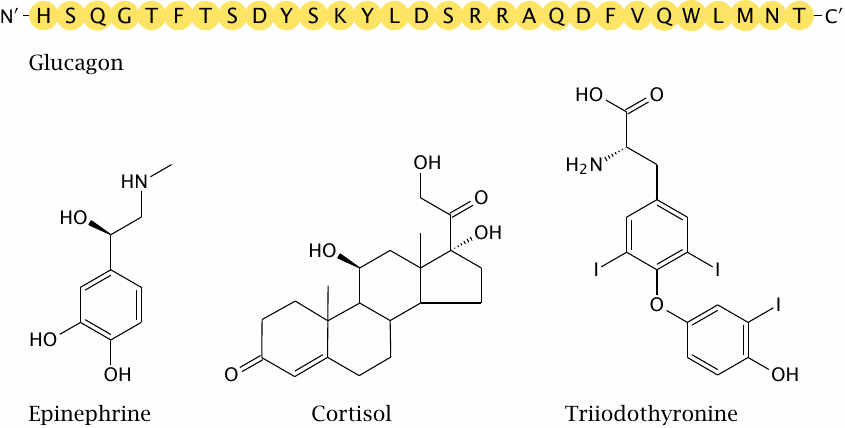
Glucagon is a peptide hormone that is produced in the α-cells of the pancreatic islets. Epinephrine is produced in the medulla, and cortisol in the cortex of the adrenal glands. All these hormones are antagonists of insulin.
| 13.3.1 |
Glucagon and epinephrine act via G-protein-coupled receptors |
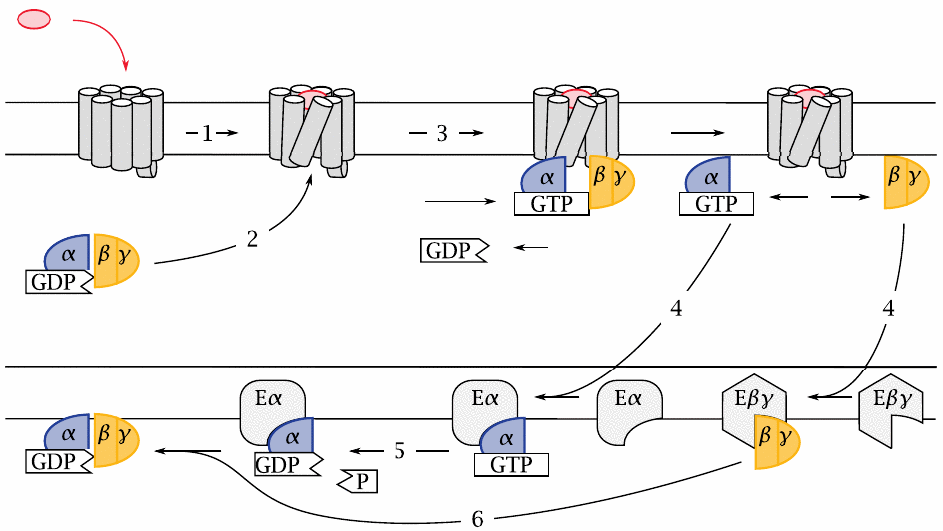
The cognate receptors of glucagon and epinephrine belong to the class of G protein-coupled receptors (GPCRs). Like receptor tyrosine kinases (RTKs), GPCRs are located in the cytoplasmic membrane. In contrast to RTKs, however, GPCRs do not have any enzymatic activity themselves. A GPCR functions solely by changing conformation in response to the binding of its cognate agonist, which occurs on the extracellular side (1). This conformational change is recognized on the intracellular side by a heterotrimeric G protein (2), which becomes bound to the receptor and is thereby activated. The active state of the G protein is stabilized by the non-covalent binding of GTP, which replaces a GDP molecule that was left behind in a previous round of activation (3). Upon GTP binding, the G protein dissociate into the α-subunit and the βγ-dimer, each of which then seeks out its cognate effector protein (4).
The α-subunit has a built-in, slow GTPase activity. When this activity kicks in and cleaves the bound GTP molecule (5), the activated state is terminated,87 and the α-subunit rejoins a βγ-dimer (6). The inactive trimer then awaits the next round of activation by the same or another GPCR molecule.
| 13.3.2 |
The glucagon and epinephrine receptors activate adenylate cyclase and protein kinase A |
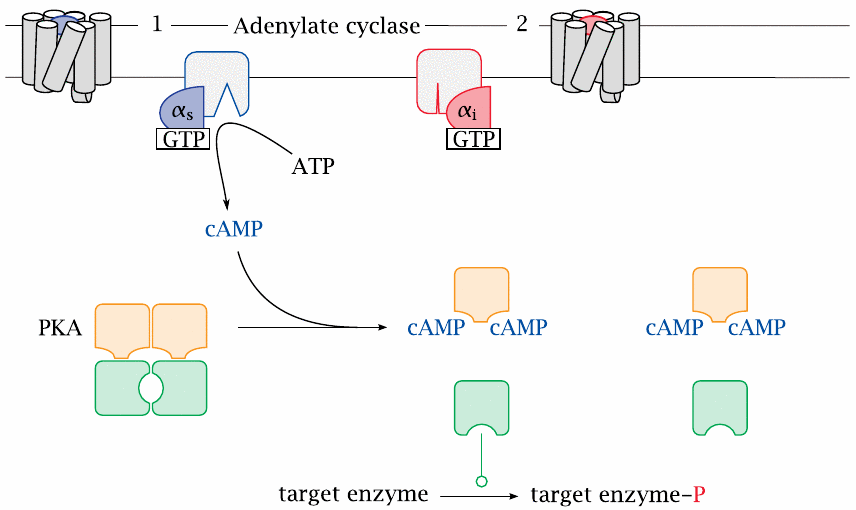
One type of G protein can couple to several types of GPCR; this is the case with the the so-called stimulatory G protein (GS), which couples to both the glucagon receptor and the β-adrenergic receptor, which binds epinephrine. The α-subunit of this G protein (αS) activates adenylate cyclase, which converts ATP to cyclic AMP (cAMP). This second messenger then binds and activates protein kinase A (PKA), which phosphorylates a number of target enzymes.
Adenylate cyclase is also targeted by the α-subunit an inhibitory G protein, Gi, which is activated downstream of other GPCRs, such as for example α2-adrenergic receptors or several serotonin receptor subtypes. Another major signaling cascade that is controlled by GPCRs is the phospholipase C/protein kinase C pathway, which is activated for example by vasopressin and oxytocin receptors. In these notes, however, we will confine the discussion to epinephrine, glucagon, and protein kinase A.
| 13.3.3 |
Metabolic effects of protein kinase A |
| Target | Effect | Metabolic consequence |
| glycogen synthase | ↓ | glucose is not locked up in glycogen, remains available |
| phosphorylase kinase | ↑ | phosphorylase is activated, glucose is released from glycogen storage |
| PFK-2 / Fructose-2,6-bisphosphatase | ↓ / ↑ | Fructose-2,6-bisphosphate drops; glycolysis is inhibited, gluconeogenesis is activated |
| hormone-sensitive lipase | ↑ | fatty acids are mobilized for β-oxidation and ketogenesis |
This slide summarizes downstream effects of PKA activation that were already discussed in detail earlier. The roles of PKA in gluconeogenesis and in glycogen synthesis are shown in slides 7.5.4 and 8.4.2, respectively. PKA also activates hormone-sensitive lipase in fat tissue, which induces the release of free fatty acids and glycerol (slide 10.3.7).
| 13.3.4 |
Glucocorticoids and thyroid hormones act on nuclear hormone receptors to activate transcription |
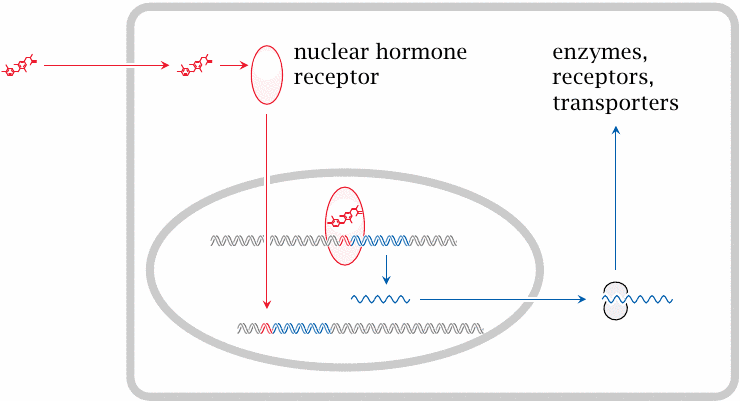
While they are inactive, nuclear hormone receptors are located in the cytosol. When activated by ligand binding, they translocate to the nucleus and bind to cognate DNA sequences, recruit a number of other regulatory proteins (not shown) and ultimately induce the transcription of genes in the vicinity of their target DNA sequences.
| 13.3.5 |
DNA binding by thyroid hormone receptors |
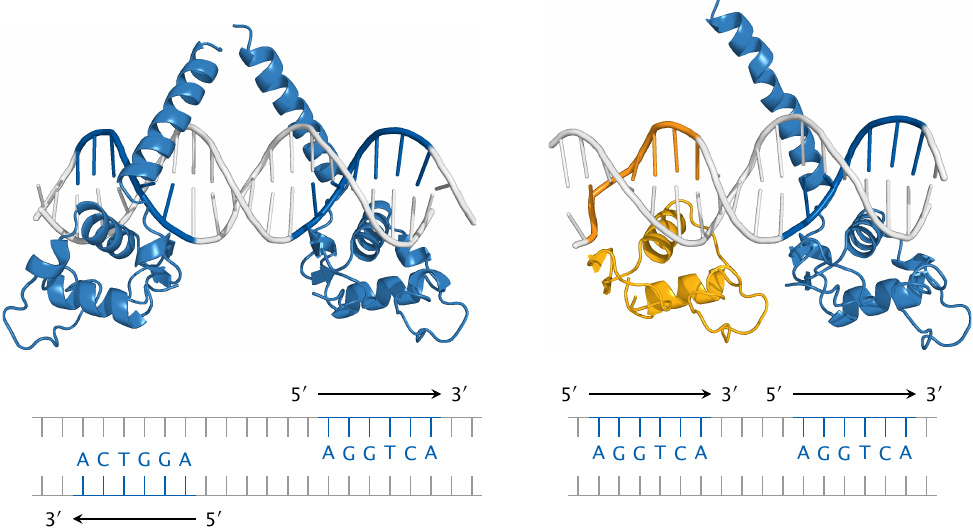
Nuclear hormone receptors function as dimeric molecules. This picture illustrates the binding of a thyroid hormone receptor homodimer (TRβ; left) and of a heterodimer of TRβ with retinoid X receptor (RXR; yellow) to specific target sequences in the DNA. The structures of the receptor molecules (rendered from 3m9e.pdb and 2nll.pdb) are not complete; the hormone-binding domains are missing, and only the DNA-binding domains are shown.
Note that TRβ and RXR bind to the same hexanucleotide motif. Nevertheless, the two receptor dimers recognize different DNA target sequences, since the two instances of the hexanucleotide differ in orientation and spacing. Therefore, the two dimers bind to different sites on the DNA and control different sets of genes.
One key mechanism through which thyroid hormones affect energy metabolism is the transcriptional induction of mitochondrial uncoupling proteins. As discussed earlier (slide 6.3.1), uncoupling proteins mediate the conversion of metabolic energy to heat and therefore increase the burn rate of glucose and other energy-rich substrates. Nevertheless, thyroid hormones do not reduce blood glucose, since they also induce the expression of β-adrenergic receptors, and they therefore amplify the glucose-enhancing effect of epinephrine.
| 13.3.6 |
Metabolic effects of glucocorticoid hormones |
- induction of enzymes for glycogen synthesis, glycogen breakdown, as well as gluconeogenesis
- induction of enzymes for protein breakdown, which supplies substrates for gluconeogenesis
- induction of adrenergic receptors
…overall, glucocorticoids increase blood glucose
In addition to their role in metabolic regulation, glucocorticoids also inhibit inflammation and immune responses. They are used as drugs in the treatment of various inflammatory and autoimmune diseases; their metabolic effects then become unwanted side effects of such therapy.
| 13.3.7 |
Glucocorticoid receptor agonists and antagonists |
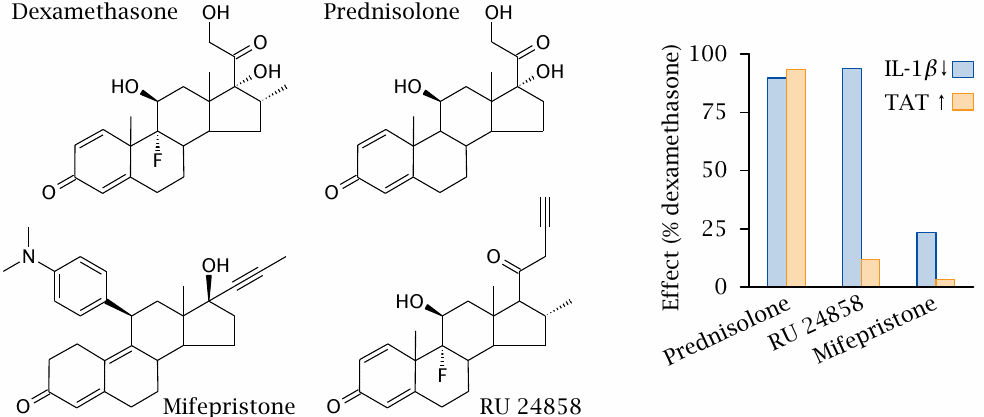
Dexamethasone and prednisolone are synthetic glucocorticoid receptor agonists that exert both antiinflammatory and metabolic effects. Mifepristone is a glucocorticoid receptor antagonist. In the experiment shown on the right, the activities of these conventional drugs were compared to those of the experimental drug RU 24858. Downregulation of interleukin-1β (IL-1β) measures antiinflammatory activity, whereas the activity of tyrosine transaminase (TAT), an enzyme that participates in amino acid degradation (see slide 12.4.5), represents metabolic regulation. Figure prepared from original data in [87].
With dexamethasone, prednisolone and mifepristone, the antiinflammatory and metabolic effects are similarly weak or strong. In contrast, with RU 24858, they are clearly distinct, suggesting that this drug selectively triggers the antiinflammatory glucocorticoid effect but avoids the side effects on metabolic regulation. Such selective glucocorticoid receptor agonists are promising but not yet in clinical use. The biochemical mechanism that underlies the dissociation of metabolic and antiinflammatory glucocorticoid effects is quite interesting [88].
Note that mifepristone is an antagonist not only at the glucocorticoid receptor but also at the progestin receptor; its typical medical applications relate to the latter activity.
| 13.3.8 |
Control of food intake by leptin |
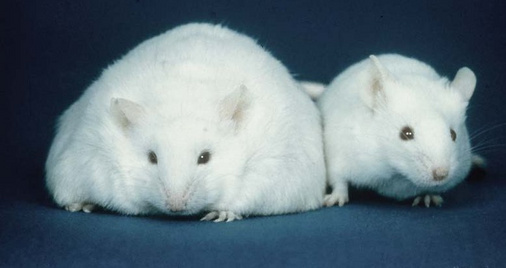
Leptin is a peptide hormone produced by fat tissue, in proportion to its abundance and current triacylglycerol content. It acts upon receptors in the hypothalamus and reduces appetite. This slide illustrates the effect of genetic leptin knock-out in mice (no brownie points for correctly identifying the knock-out and the wild-type mouse).
While leptin is important in long-term regulation of metabolism and body weight, initial expectations that leptin substitution or leptin receptor agonists might be useful in the treatment of obesity in humans have not been fulfilled. It appears that in adipose patients there is no leptin deficiency, but rather an insensitivity to it, similar to the insensitivity to insulin in type 2 diabetes (see next chapter).