| 12 |
Cancer chemotherapy |
| 12.1 |
Overview |
The purpose of cancer chemotherapy is analogous to that of antimicrobial chemotherapy, namely, to selectively kill the pathogenic cells. Of the two, cancer chemotherapy tends to be more toxic, yet less reliably effective; or in other words, it fails to achieve the level of selective toxicity that is possible with antimicrobial chemotherapy. This is not surprising, since the cells that we aim to destroy originate in our own bodies, and therefore contain few or no drug targets that are truly distinct from those found in healthy cells. Considering this lack of molecular distinctiveness, it is rather remarkable that cancer chemotherapy even works as well as it does.
Like pathogenic microbes, cancer cells may develop resistance to anticancer drugs under therapy. To mitigate this problem, anticancer drugs are almost always used in combination.
| 12.1.1 |
Forms of cancer therapy |
- Surgery
- Radiation
- Chemotherapy
Criteria for therapy selection
- Benign or malignant tumor
- Tissue of origin, histological variant of tumor
- Stage of tumor—early and localized vs. advanced and disseminated
Broadly speaking, surgery is applied locally, radiation regionally, and chemotherapy systemically.
Benign tumors don’t infiltrate the surrounding tissue and don’t give rise to metastases; therefore, they can usually be cured through surgery alone. In most solid malignant tumors, surgery is used to remove the main tumor mass as far as possible. Subsequently, radiation or chemotherapy are applied alone or together in order to extirpate any regionally or systemically disseminated tumor cells and thus prevent resurgence of the cancer. However, leukemias and lymphomas, as well as some specific solid tumors, are treated primarily with chemotherapy.
As we had seen in Chapter 7, the cells in some of our organs depend on growth stimulation by hormones. Tumor cells originating in these organs may retain this hormone dependency; important examples are breast and prostate cancers, which often remain dependent on growth stimulation by sexual steroid hormones. In such cases, withdrawal of the hormonal stimulus, using receptor antagonists or inhibitors of hormone synthesis, can be an effective part of chemotherapy. Where such specific hormone dependency does not exist, broadly cytotoxic drugs that interfere with mitosis, DNA synthesis, or other fundamental aspects of cell biology are often the only option. In any case, such cytotoxic drugs are part of most combination regimens.96
It is important to understand that cytotoxic drugs don’t simply disrupt and destroy the cancer cells with brute force. Instead, they trigger regulatory pathways within the cell that initiate programmed cell death, or apoptosis. Accordingly, mutations that inactivate apoptotic pathways are often responsible for resistance of tumor cells to chemotherapy.
| 12.2 |
Cellular pathways that control proliferation and apoptosis |
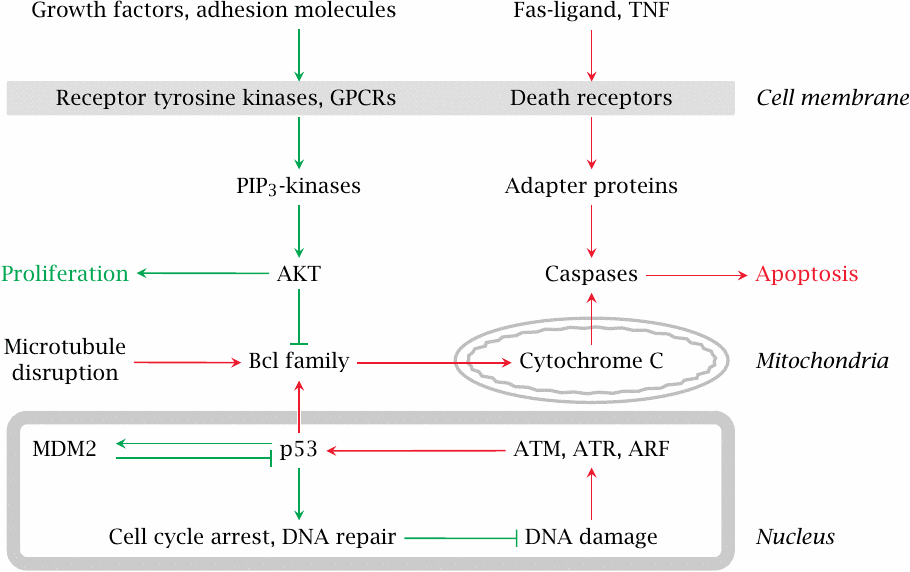
The lifetimes and rates of proliferation of the cells in our bodies are highly variable. Most nerve cells in the brain don’t ever divide but just stay put throughout life,97 while those in the bone marrow and the intestinal epithelia proliferate rapidly; lymphocytes often end their days through apoptosis, which is necessary to avoid damage to the body through autoimmune reactivity. The pathways that determine whether a cell should persist, proliferate, or commit apoptosis are subject to both intra- and extracellular signals.
Growth factors and adhesion molecules stimulate the PIP3 kinase pathway and thereby stimulate proliferation and inhibit apoptosis. Conversely, mediator proteins such as Fas-Ligand and tumor necrosis factor (TNF) act on so-called “death receptor” proteins in the cell membrane. This leads to the activation of caspases, a family of cysteine proteases that cleave their numerous substrates at aspartate residues, and thereby destroy the cell.
In the intrinsic pathway of apoptosis, DNA damage leads to the activation of protein kinases such as ATM and ATR, which in turn activate the transcriptional regulator p53. Subsequently, increased expression of certain regulatory proteins in the Bcl family leads to the permeabilization of mitochondria by Bax, another Bcl family member. Cytochrome C released from the mitochondria then again activates caspases.
Mutations in many of the proteins shown here have been implicated in the induction or progression of cancer. One such protein, p53, is disabled by mutation in as many as 50% of all malignant tumors, which highlights its central role in tumor suppression.
| 12.2.1 |
Dysregulation of growth in tumor cells |
Normal body cells
- grow or persist only when stimulated by growth factors
- undergo apoptosis when deprived of growth factor stimulation
Tumor cells contain mutations that
-
create surrogate growth stimuli
- constitutively active growth factor receptors
- autocrine secretion of growth factors
- disrupt activation of apoptosis downstream of growth factor deprivation
- but also make tumor cells more susceptible to some apoptotic stimuli than normal cells
It was noted above that p53 is very frequently inactivated in malignant tumors. This protein has an ambivalent role: while it promotes apoptosis when maximally activated, at lower degrees of activation it can actually promote cell survival. It does this by initiating a pause in cell division and activating DNA repair systems. Failure to pause division and perform DNA repair may be one of the reasons why tumor cells actually tend to be more susceptible to certain apoptotic stimuli than normal cells. On the other hand, failure of DNA repair also increases the mutation rate, which in turn accelerates the progression of the tumor to more malignant and invasive behavior, as well as the development of resistance to chemotherapy.
The readiness to plunge into apoptosis varies not only between normal cells on one hand and tumor cells on the other, but also between normal cells from different tissues. For example, lymphocytes and their precursors are driven into apoptosis extremely easily (cf. slide 10.3.2). Tumors derived from lymphocytes—lymphomas and lymphatic leukemias—often retain this trait, and therefore are much more amenable to chemotherapy than most other tumors. On the other hand, tumors derived from normal cells that have little inclination to commit apoptosis may inherit this trait also, and therefore tend to be quite impervious to chemotherapy. Examples are cancers originating in the brain and the kidneys.
| 12.2.2 |
Cellular models of dysregulated apoptosis |
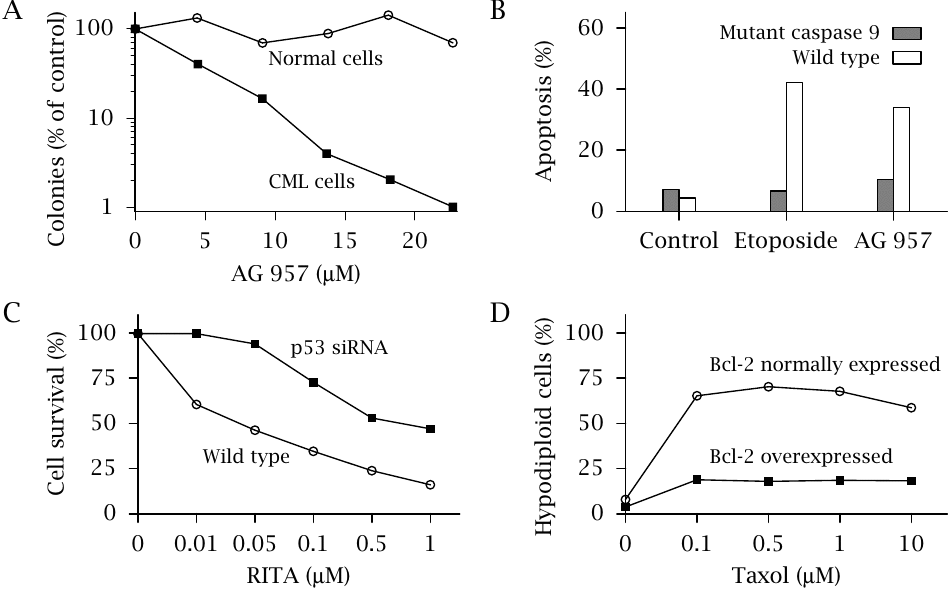
This slide shows several experiments that illustrate some of the points made above on the dysregulation of apoptosis in tumors.
- (A)Apoptotic response triggered by the tyrosine kinase inhibitor AG 957 in white blood cells isolated from a patient with chronic myeloic leukemia (CML) and in normal control cells. This drug inhibits receptor tyrosine kinases, including a mutant one that is constitutively active and sustains the proliferation of the malignant cells. Inhibition of this type of protein kinase causes apoptosis more readily in the malignant cells than in comparable normal ones.
- (B)Expression of a mutant of caspase 9 in a leukemic cell line (K562) inhibits apoptosis in response to two anticancer drugs, namely the topoisomerase II inhibitor etoposide as well as AG-957. Caspase 9 is part of the destructive machinery that is activated downstream of different proapoptotic stimuli (see slide 12.2). In some tumors, mutants of the enzyme may be expressed that not only are inactive themselves but even suppress the activity of simultaneously expressed active protease; that is, they are dominant negative. As expected, such mutants inhibit the induction of apoptosis by anticancer drugs.
- (C)The experimental compound RITA98 inhibits the binding of the inhibitory protein MDM2 to p53 (see slide 12.2), thus increasing the proapoptotic activity of p53. Inhibiting the expression of p53 with a specific siRNA in a model cell line reduces apoptosis in response to this drug.
- (D)The drug taxol disrupts microtubules, which indirectly leads to the activation of pro-apoptotic proteins in the Bcl family. Overexpression of the inhibitory regulator protein Bcl-2 reduces apoptosis, quantified here by FACS counting of “hypodiploid cells,” which are not intact cells but rather fragments of nuclei released from apoptotic cells.
The plots in this slide were redrawn from data found in various sources [117–119].
| 12.3 |
The cell cycle and its checkpoints |
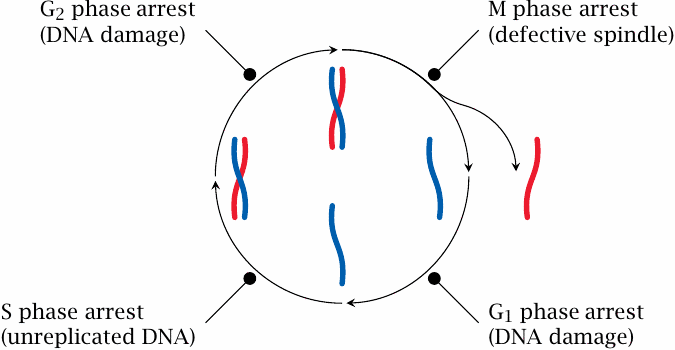
Somatic cell proliferation goes through successive coordinated phases, which together constitute the cell cycle. Daughter cells arise by mitosis, which within the cell cycle is referred to as the M phase. After the G1 phase,99 a cell prepares for the next round of division by entering the S phase, in which the DNA is replicated, that is, a second double-stranded DNA copy is synthesized for each chromosome. Following another short intermission, the G2 phase, mitosis repeats.
In each of these phases, corresponding checkpoint proteins are activated that will arrest the cell cycle if they detect phase-specific forms of genetic damage. Depending on the extent of damage, the arrest may be followed by DNA repair and resumption of the cell cycle, or apoptosis may be triggered.
In the M phase, cell cycle arrest may be induced if defects are found in the mitotic spindle, that is, the cytoskeletal apparatus that distributes the chromosomes evenly between the two daughter cells. Defects in the M phase checkpoint proteins will permit the survival of cells that have gained or lost chromosome fragments, entire chromosomes, or even acquired extra copies of the whole genome. Cells whose chromosome complement deviates from the regular diploid one are called aneuploid. The occurrence of aneuploid cells is a hallmark of cancer and contributes greatly to genetic variability and development of chemoresistance.
| 12.3.1 |
Progressive cell aneuploidy in a recurring tumor |
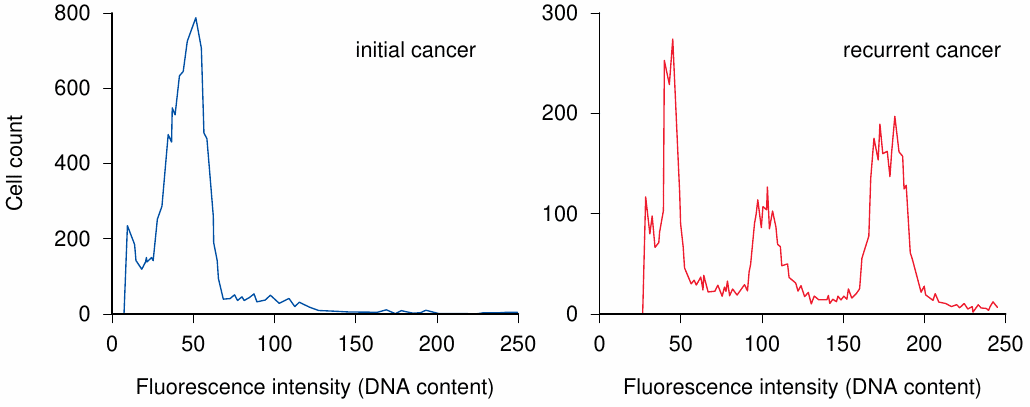
In the experiment shown, the nuclei of cells obtained from a sarcoma, that is, a malignant tumor derived from a non-epithelial tissue, were isolated and incubated with a fluorescent dye such as propidium iodide, which emits fluorescence only after intercalation into DNA. The stained nuclei were then passed through a flow cytometer. As the nuclei pass the instrument’s laser beam in single file, each nucleus causes a fluorescence pulse, the intensity of which is proportional to its DNA content.
In the original tumor (A), the DNA content per cell shows one major peak, corresponding to the diploid chromosome complement. In the recurring tumor (B), there are multiple peaks, indicating aneuploidy and clonal divergence.
Aneuploidy is very common with malignant tumors. The additional or missing gene copies will change the expression levels of individual proteins, including ones that influence tumor proliferation, apoptosis, and susceptibility or resistance to anticancer drugs. From the genetically heterogeneous cell population, particularly resistant cell clones may be selected under drug therapy. Figure prepared from original data in [120].
| 12.4 |
Cell type-specific anticancer drugs |
- Hormones and growth factors: interferon-α in hairy cell leukemia
- Hormone antagonists: most significant with breast and prostate cancer
- Tissue-specific prodrug activation: mitotane in adrenal gland tumors
- Tissue-specific accumulation of radioactive iodine: thyroid cancer
Among the numerous anticancer drugs, we can broadly distinguish those that act only on specific cell types from those that are generally cytotoxic. We will now consider some examples of drugs in the first category.
| 12.4.1 |
Sexual hormones and receptor antagonists |
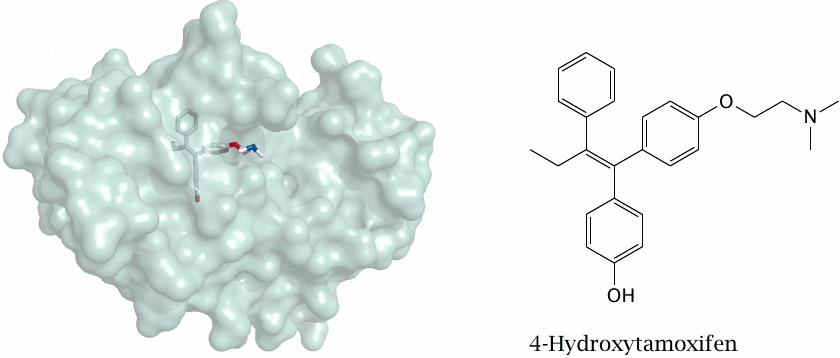
Estrogens like estradiol or progestins like progesteron are required by many breast or uterine cancers, which can therefore be treated with estrogen receptor antagonists like tamoxifen or progestin receptor antagonists like mifepristone. Similarly, androgen-dependent prostate cancers can be targeted with inhibitors testosterone-5α-reductase or androgen receptor antagonists (see slide 7.4.9).
The structure (rendered from 2p7z.pdb) in this slide shows 4-hydroxy-tamoxifen, an active metabolite of tamoxifen, bound to the ligand-binding domain of the estrogen receptor; the formula of the free compound is rendered in a similar orientation. Mifepristone is shown in slide 7.4.5.
| 12.4.2 |
Aromatase and two of its inhibitors |
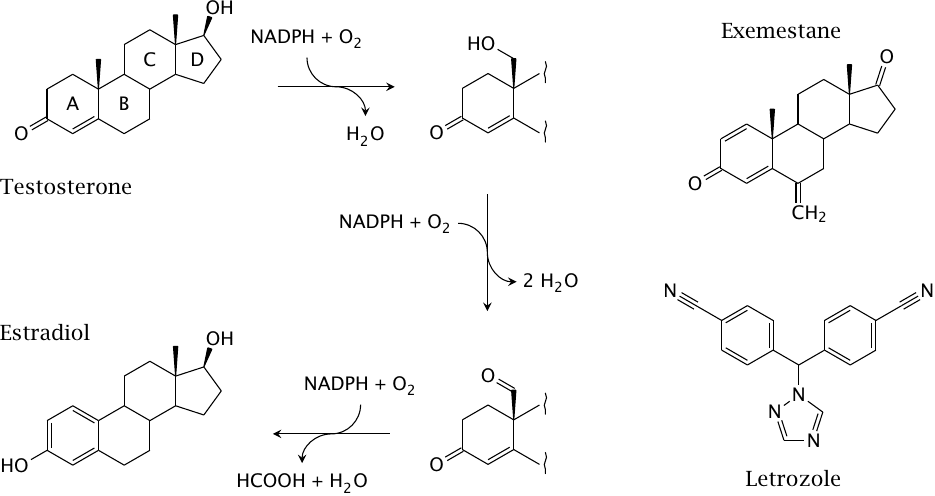
Aromatase is a cytochrome P450 enzyme that converts androgens to estrogens (see slide 7.4.8). The left part of this slide shows details of the aromatase reaction. The methyl group between rings A and B is converted in two steps to an aldehyde. The keto group in ring A then becomes its enol tautomer, which creates a second double bond in the ring. The third double bond is introduced into ring A concomitantly with oxidative cleavage of the exocyclic aldehyde as formic acid.
The slide also shows the structures of the clinically used aromatase inhibitors exemestane and letrozole. Exemestane is a steroidal, covalent aromatase inhibitor. The exocyclic C=C double bond likely reacts with a nucleophile in the active site [121]. Letrozole is a nonsteroidal, noncovalent aromatase inhibitor. It is somewhat similar in structure to the antifungal drug miconazole, an inhibitor of 14-α-sterol demethylase (see section 11.8.2) that, like aromatase, belongs to the cytochrome P450 family.
| 12.4.3 |
The anticancer prodrug mitotane is selectively activated in the adrenal cortex |
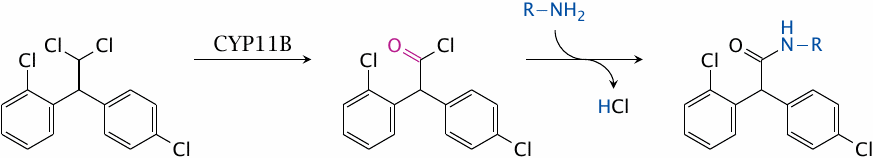
Mitotane is converted to a reactive acyl chloride by 11-β-hydroxylase, a cytochrome P450 variant (CYP11B). The acyl chloride then reacts with amino groups or other nucleophiles in proteins or nucleic acids.
11-β-Hydroxylase participates in the synthesis of cortisol from progesterone in the cortex of the adrenal glands (see slide 7.4.3). It is highly expressed only in this tissue, which is therefore selectively susceptible to mitotane. Adrenal gland tumors that retain the expression of this enzyme can be treated with mitotane.
| 12.5 |
Anticancer drugs that target specific oncoproteins |
- α-Retinoic acid in promyelocyte leukemia
- Imatinib in chronic myeloic leukemia; other tyrosine kinase inhibitors in various tumors
- Monoclonal antibodies against growth factor receptors on the cell surface, for example Her2/neu (“herceptin”) in breast cancer
Oncoproteins are mutant proteins that drive the proliferation of aberrant cells in tumors. Key examples are the oncoproteins involved in promyelocyte leukemia and in chronic myeloic leukemia, which both arise from chromosome translocations; this is discussed in more detail below.
The growth factor receptor Her2/neu is an oncoprotein found in a large number of breast cancers and also in some other carcinomas. Mutations affecting Her2/neu function found in tumors typically do not change the structure of the protein as such, but instead cause it to be overexpressed or excessively activated by phosphorylation.
| 12.5.1 |
A chromosomal translocation causes promyelocytic leukemia |
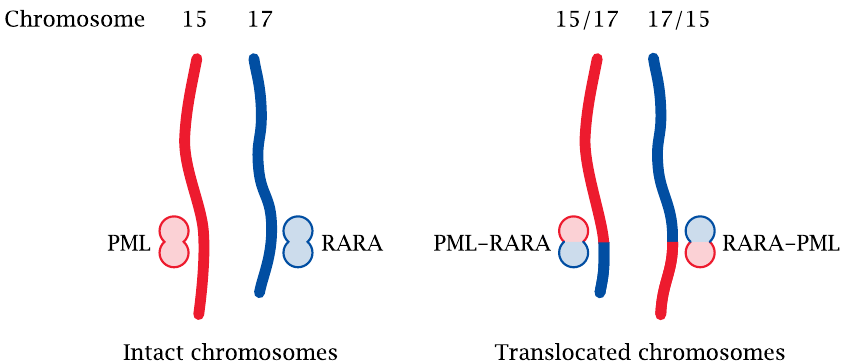
Bone marrow stem cells give rise to mature blood cells in several successive stages of proliferation and differentiation. In all stages except the final ones, individual cells can either divide and produce like daughter cells, or advance to the next stage of differentiation.
One of the intermediate stages in granulocyte differentiation is the promyelocyte. In promyelocytic leukemia (PML), this cell type proliferates excessively and displaces the regular bone marrow, which causes shortage of all sorts of normal blood cells. PML is caused by a specific chromosomal translocation, that is, the reciprocal exchange of chromosome fragments between chromosomes 15 and 17.
Two genes, respectively named PML and RARA, span the fault lines of the translocation on chromosome 15 and 17, respectively. Translocation gives rise to two chimeric genes, which are referred to as RARA-PML and PML-RARA. Of the two gene products, PML-RARA is the one that functions as an oncoprotein.
| 12.5.2 |
The mutant PML-RARA receptor blocks promyelocyte differentiation |
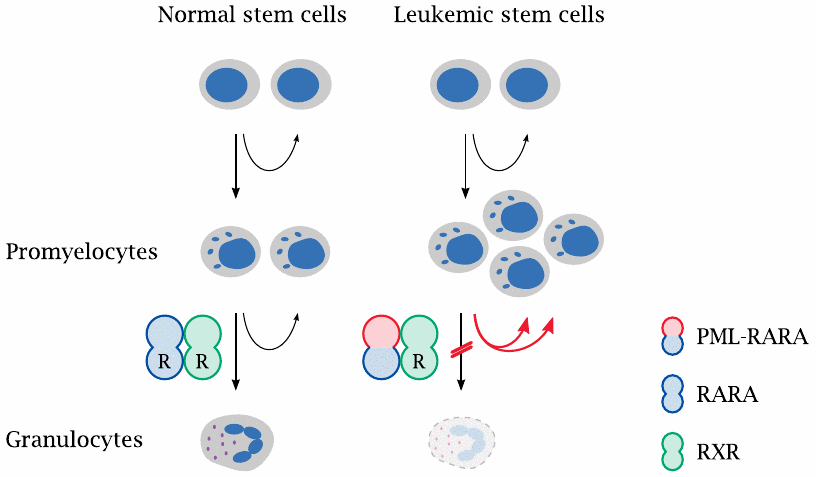
PML-RARA is an in-frame fusion between the N-terminal portion of the PML gene product and the C-terminal part of the RARA gene product, which is the retinoic acid receptor α. White blood precursor cells that express this protein can proliferate and progress through the early stages of differentiation seemingly without disturbance.
Differentiation stalls, however, at the promyelocyte stage. The progression to the next (the myelocyte) stage requires the α-retinoic acid receptor to form a functional heterodimer with another retinoid acid-binding receptor, the retinoid X receptor (RXR).100 PML-RARA still forms heterodimers with RXR. However, at normal levels of retinoic acid, PML-RARA fails to bind its ligand, and in this form the receptor dimer engages in aberrant transcriptional regulation that causes cell differentiation to stall. Dysfunctional promyelocytes accumulate and proliferate, thereby displacing normal blood cell formation.
| 12.5.3 |
Retinoic acid therapy restores promyelocyte differentiation |
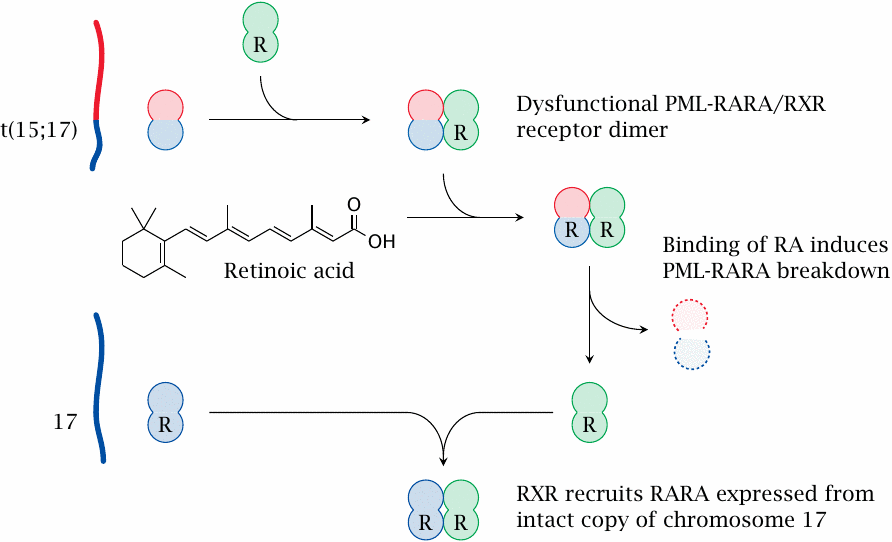
Fortunately, in most patients,101 this condition can be treated with high dosages of retinoic acid. While PML-RARA has a reduced affinity for retinoic acid, this can be overcome by applying it as a drug at high concentrations. This changes the regulatory activity of the receptor dimer and also drives the drug-bound PML-RARA protein into accelerated degradation. With the aberrant protein gone, RXR can combine with intact RARA that continues to be expressed from the intact copy of chromosome 17; cell differentiation resumes, and excessive promyelocyte proliferation ceases.102
| 12.5.4 |
Chronic myeloid leukemia |
- Caused by reciprocal translocation between chromosomes 9 and 22 (“Philadelphia chromosome”)
- Translocation produces a chimeric, constitutively active protein tyrosine kinase (Bcr-Abl) that drives the proliferation of myeloid precursor cells
- Treatment with imatinib or other tyrosine kinase inhibitors controls, but usually does not eradicate leukemic cells
- After several years, CML typically ends in a “blast crisis”, which resembles an acute myeloid leukemia
The two stages of chronic myeloid leukemia (CML) are somewhat resemblant of a solid tumor transitioning from benign to malignant growth. In contrast to such a benign tumor, CML grows diffusely and in multiple locations right away; however, this may under certain circumstances be observed with normal bone marrow tissue also. As in a benign tumor, the growth-dysregulated cells in early-stage CML remain genetically homogeneous, and their growth can be suppressed by inhibiting a single oncoprotein.
Like the acute myeloic leukemia it resembles, the terminal blast crisis is treated with combinations of multiple cytotoxic drugs. We will see examples of such drugs a little further on this chapter.
| 12.5.5 |
Imatinib bound to its target enzyme c-Abl kinase |
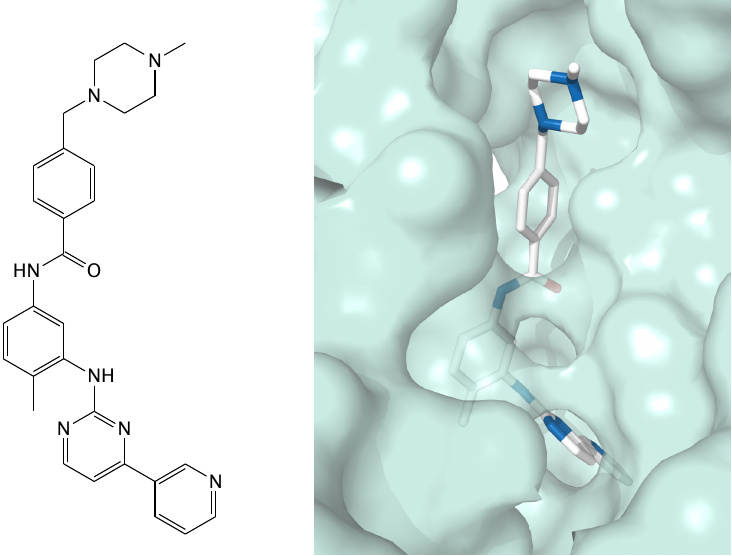
The mutant Bcr-Abl kinase that induces CML inherits the tyrosine kinase domain from the normal cellular kinase c-Abl, which is the protein that was used in this crystallographic experiment.
The structural formula of imatinib is oriented similarly to the conformation of the drug molecule inside the enzyme. Structure rendered from 1m52.pdb.
| 12.6 |
Cytotoxic anticancer drugs |
- Antimetabolites
- Inhibitors of DNA topoisomerase
- Proteasome inhibitors
- Inhibitors of mitosis
- DNA-alkylating and other DNA-damaging agents
Cytotoxic anticancer drugs have multiple different targets and modes of action, but a common motif is that they inflict damage on the cells in order to activate the intrinsic pathway of apoptosis. While different cell types may respond more or less readily to such treatment, the apoptotic pathway itself is functional in all cells, and it is usually not possible to limit the cytotoxic effect to specific cell types.
We will now look at selected examples for the different types of cytotoxic anticancer drugs, starting with the antimetabolite 5-fluorouracil.
| 12.6.1 |
Dual action mode of 5-fluorouracil |
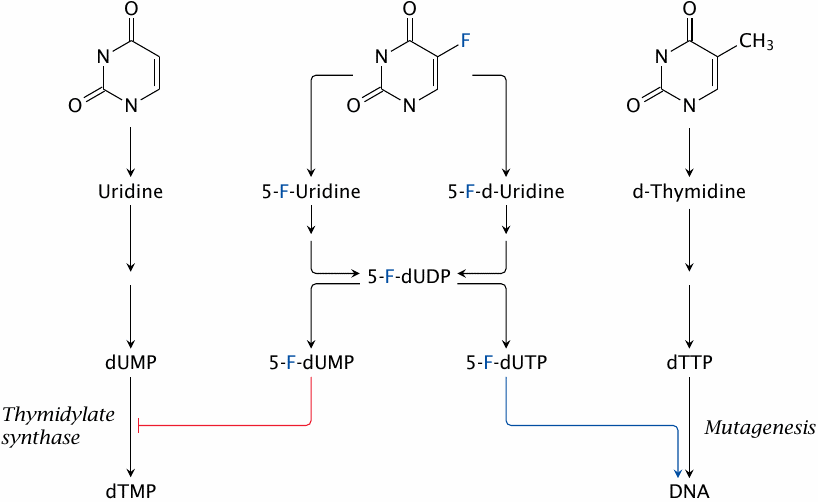
5-fluorouracil (5-FU) is a base analog that mimics both uracil and thymine; the dual resemblance gives rise to a twofold mode of metabolic activation and of action. One of the initial activation products, 5-fluorodeoxyuridine, is also used as a drug itself.
The metabolic activation of 5-FU occurs along the so-called salvage pathways that recycle bases and nucleosides released by nucleic acid degradation. 5-FU is activated by the same enzymes that salvage uracil or thymine and is thereby converted to 5-FdUMP, which is a suicide substrate for thymidylate synthase (see next slide). This is its major mode of action and the one that makes it an antimetabolite.
The precursor of 5-FdUMP is the diphosphate (5-Fluoro-dUDP). Due to the resemblance between 5-FU and thymine, 5-Fluoro-dUDP may also be further phosphorylated to to 5-Fluoro-dUTP, which is incorporated into DNA; this will cause point mutations (see slide 12.6.3).
| 12.6.2 |
Catalytic mechanism of thymidylate synthase |
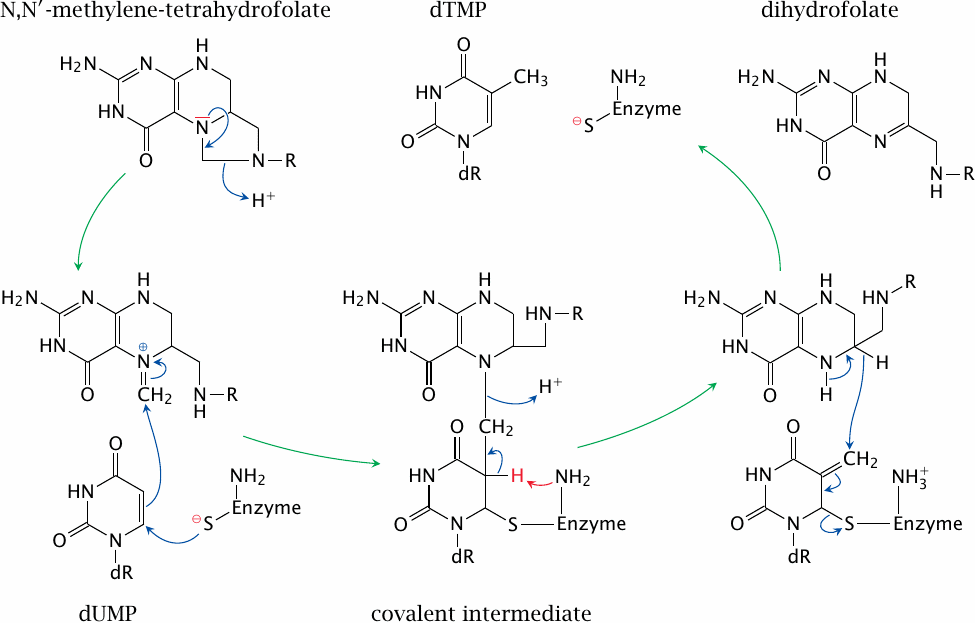
Thymidylate synthase attaches a methyl group onto uracil to form thymine, at the level of the deoxyriboside monophosphate. It acquires the methyl group from the cosubstrate N,N′-methylene-tetrahydrofolate, which is thereby converted to dihydrofolate.
Halfway through the reaction, the enzyme, the substrate and the cosubstrate are all covalently connected. Resolution of this covalent intermediate depends on the abstraction of a hydrogen atom from position 5 of uracil (highlighted in red). In 5-FU, the place of this hydrogen is taken by fluorine. The fluorine will resist abstraction, the complex will fail to resolve, and the enzyme will remain covalently trapped and inactivated. The resulting lack of dTMP and dTTP inhibits DNA replication.
| 12.6.3 |
Mutagenesis through mispairing of the 5-FU iminol tautomer |
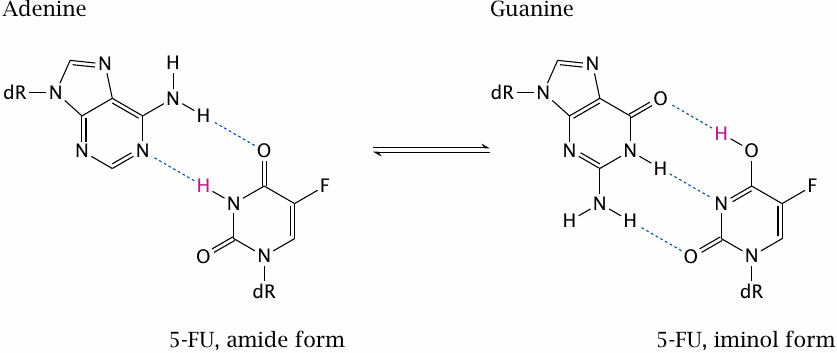
5-F-dUTP can be incorporated into DNA and promote point mutations. This is due to the fluorine, which withdraws electrons from the ring; this, in turn, pulls electrons into the ring at other positions and encourages the molecule to assume the iminol configuration. In this configuration, the base no longer pairs with adenine but with guanine.
If the iminol configuration is present during DNA replication, guanine will be selected and incorporated into the opposite strand. This mutagenic effect of 5-FU augments its anticancer effect.
| 12.6.4 |
Thymine and various halogen analogues |
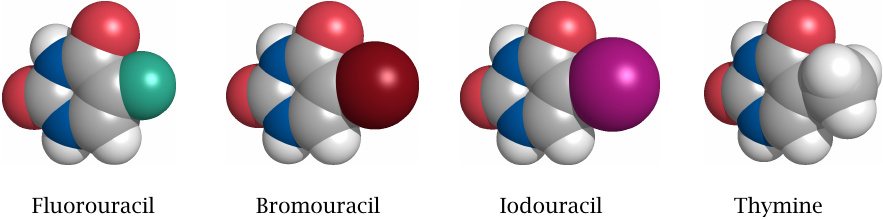
The bromo- and iodo-analogs of deoxythymidine, 5-bromouracil-deoxyriboside (5-BUdR) and idoxuridine, are also incorporated into DNA. Bromine and iodine are larger than fluorine and similar to a methyl group in size. Therefore, the activated triphosphates more closely resemble dTTP and are more effectively incorporated into DNA than the 5-FU derivative.
Like fluorouracil, idoxuridine is used in tumor therapy. 5-BUdR is not used clinically but has been widely used for shotgun mutagenesis experiments in genetic research.
| 12.6.5 |
Blockade of dihydrofolate reductase also inhibits thymidylate synthesis |
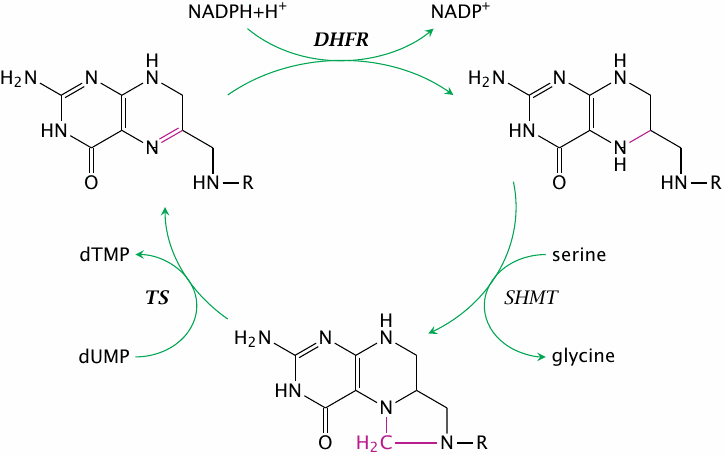
As we have seen in slide 12.6.2, thymidylate acquires a methyl group from N,N′-tetrahydrofolate, which is thereby turned into dihydrofolate. Recycling dihydrofolate involves two successive enzyme reactions, namely the reduction to tetrahydrofolate, followed by the acquisition of a methylene group from serine hydroxymethyl transferase (SHMT) or from the glycine cleavage system. If dihydrofolate reductase (DHFR) is inhibited, this will indirectly also inhibit the synthesis of dTMP by thymidylate synthase (TS).
In contrast to 5-FU, inhibitors of dihydrofolate reductase do not resemble nucleotides and will not directly interact with DNA to cause mutations. Therefore, they are less toxic and are used not only in cancer but also for long-term treatment in autoimmune diseases such as lupus erythematosus or myasthenia gravis.103
| 12.6.6 |
Inhibitors of dihydrofolate reductase |
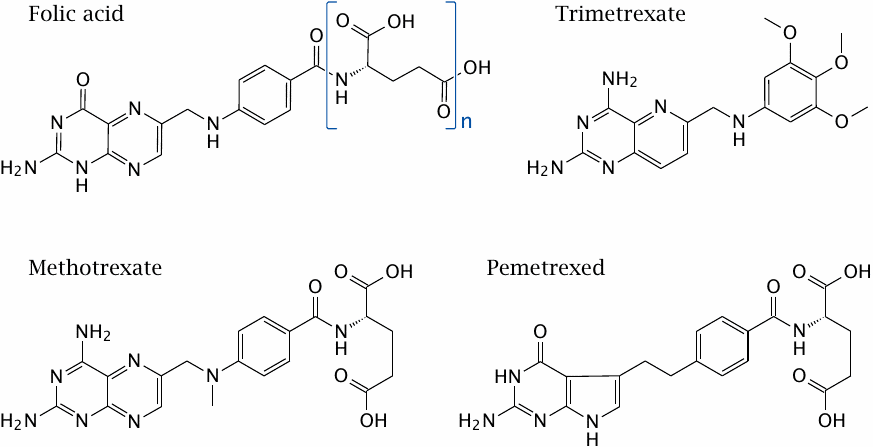
Methotrexate and pemetrexed inhibit the human dihydrofolate reductase enzyme (DHFR). Pemetrexed also inhibits thymidylate synthase directly, as well as glycinamide ribotide formyltransferase, a THF-dependent enzyme from the purine nucleotide biosynthesis pathway. Since it acts on multiple targets, it is less prone to resistance through DHFR gene amplification and overexpression, which is a common resistance mechanism with drugs that inhibit this enzyme exclusively.
Trimetrexate inhibits DHFR not only in humans but also in some microbes, and it can be used for example in the treatment of infections with the fungus Pneumocystis carinii.
In this slide, the brackets in the structure of folic acid represent a polyglutamate moiety that is attached to folic acid in human metabolism; it is involved in active transport of folate in the human body. Methotrexate and pemetrexed retain a single glutamate residue that is extended into a polyglutamate tail inside the body also. In contrast, trimetrexate does away with this polyglutamate and crosses cell membranes on its own by passive diffusion. This feature likely is important for its activity against not only human cells but also microbes.
| 12.6.7 |
Structure of cytosine arabinoside (araC) |
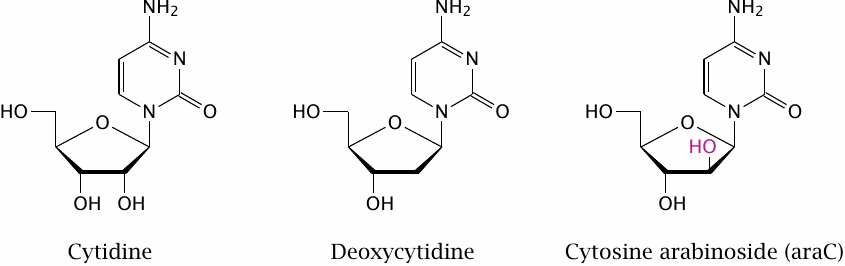
In the last chapter, we had seen some examples of nucleotide analogues that inhibit virus replication through interfering with nucleic acid polymerization (see slide 11.10.5). The same approach is used with human DNA polymerases for cancer chemotherapy.104
An example of a base analogue that interferes with human DNA polymerases is cytosine arabinoside (araC). This molecule contains arabinose, an epimer of ribose, instead of ribose or deoxyribose itself. The hydroxyl group in position 2 of this sugar points “up” rather than “down” as it does in ribose. Interference with DNA synthesis is due to this aberrant hydroxyl group.
| 12.6.8 |
Metabolic activation and inactivation of araC |
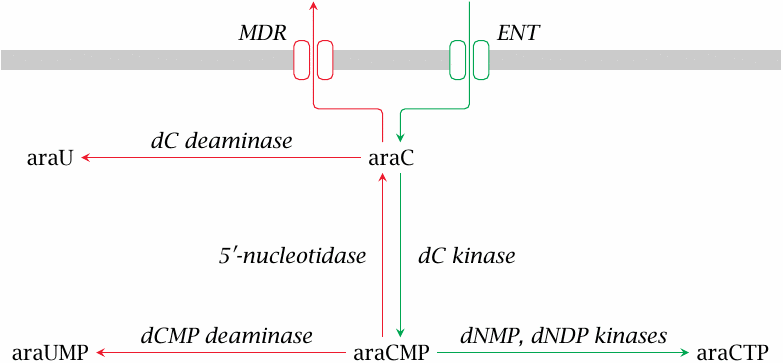
AraC enters the cell by facilitated diffusion through the equilibrative nucleoside transporter (ENT). In order to become a substrate for DNA polymerase, araC must be phosphorylated to the triphosphate (araCTP); this is accomplished by enzymes of the nucleoside salvage pathways.
The activation of araC can be reversed at several stages. Extrusion of araC itself is mediated by multi-drug resistance transporters (MDR) such as P-glycoprotein (see slide 3.5.1). Like cytidine and deoxycytidine, araC can undergo deamination either as a free nucleoside or as a monophosphate, and the initial phosphorylation can be reversed by the enzyme 5′-nucleotidase. Increased expression of MDR or of enzymes that counteract the activation of araC to araCTP cause tumor cell resistance (see slide 12.6.9).
| 12.6.9 |
Overexpression of 5′-nucleotidase in leukemic cells correlates with reduced survival rates |
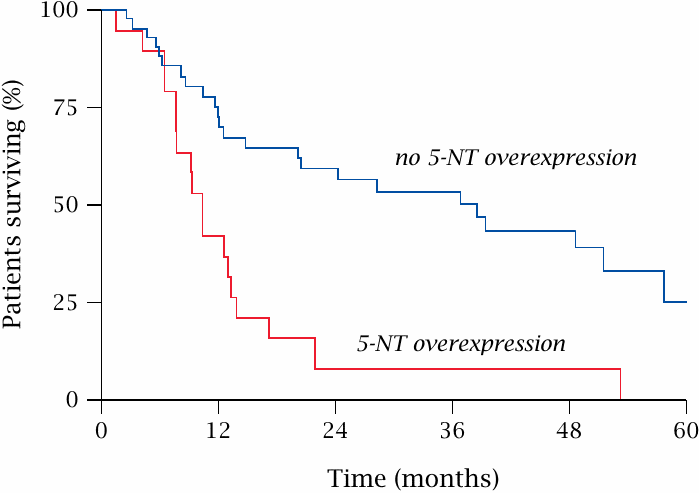
AraC is an important drug in the treatment of acute myeloic leukemia (AML). Reduced susceptibility to araC due to increased expression of 5′-nucleotidase correlates with a significant shortening of relapse-free survival in AML patients. Figure prepared from original data in [123].
| 12.6.10 |
Action mode of araCTP |
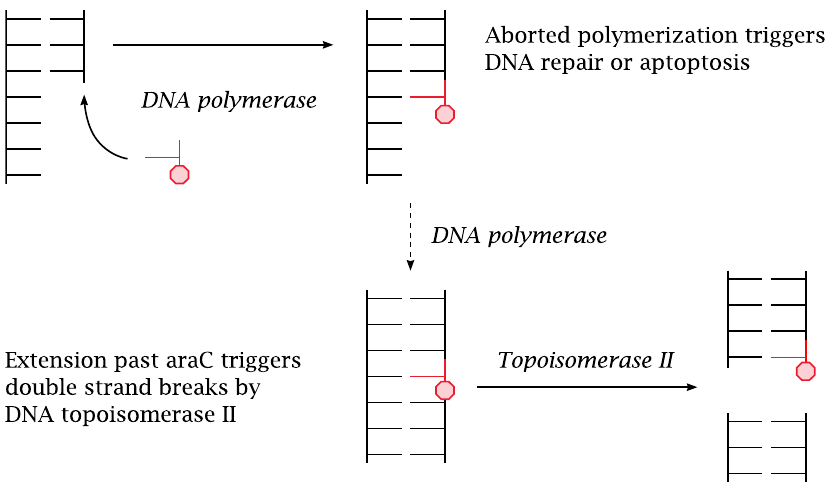
AraCTP is accepted as a substrate by DNA polymerase and becomes incorporated into a growing strand of DNA. The polymerase may or may not manage to continue past an incorporated araC molecule. If not, DNA repair will be activated, and the base will be excised and replaced; this amounts to a delay of DNA synthesis and constitutes a proapoptotic signal.
If DNA polymerase manages to continue past an incorporated araC molecule, the latter becomes part of a continuous DNA double strand. Such interposed araC residues then become preferred substrate sites for DNA topoisomerase II, which cleaves the DNA double strand [124]. Resealing of the double strand may fail and lead to chromosome breaks.
| 12.6.11 |
Function of DNA topoisomerases |
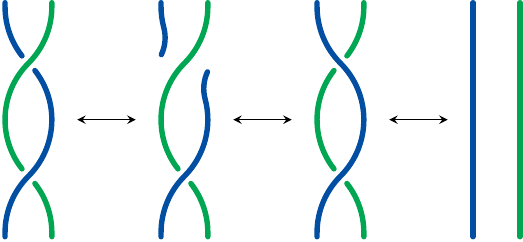
DNA topoisomerases introduce coils and super-coils into DNA molecules or remove them. They do so by breaking and rejoining the DNA molecules. By transiently “straightening out” the DNA molecules, they make them accessible to replication and transcription.
DNA topoisomerase I breaks one strand of a DNA molecule, swivels the ends around the other strand and then re-ligates them. It thus changes the number of coils in an individual double-stranded DNA molecule. DNA topoisomerase II operates analogously on two stretches of double-stranded DNA that are twisted around one another, breaking both strands of one double helix and pushing the other through the gap before rejoining the first one.
| 12.6.12 |
DNA topoisomerase inhibitors |
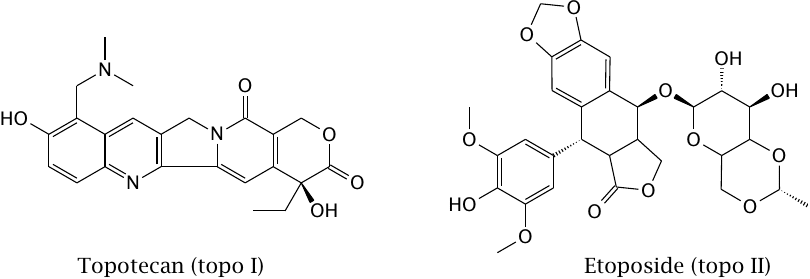
As these two examples show, inhibitors of both topoisomerase I and II are used in cancer chemotherapy. The effect of topotecan on its target is illustrated in the next slide.
| 12.6.13 |
Topotecan bound to topoisomerase I and DNA |
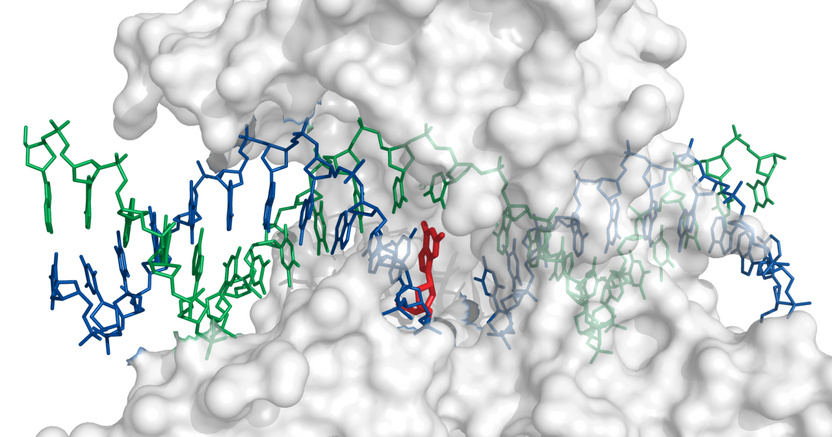
The drug (shown in red) binds to the enzyme such that it inserts itself between the free ends of the cleaved DNA strand and thereby prevents re-ligation. Structure rendered from 1k4t.pdb.
| 12.6.14 |
Bortezomib inhibits the proteasome |

Proteasomes are drum-shaped, large protease complexes. They unfold and cleave intracellular proteins destined for degradation.105 Bortezomib binds and inhibits the protease active sites in proteasomes. This causes all manner of dysregulation [125] and again promotes apoptosis.
| 12.6.15 |
Vinblastine inhibits tubulin polymerization |
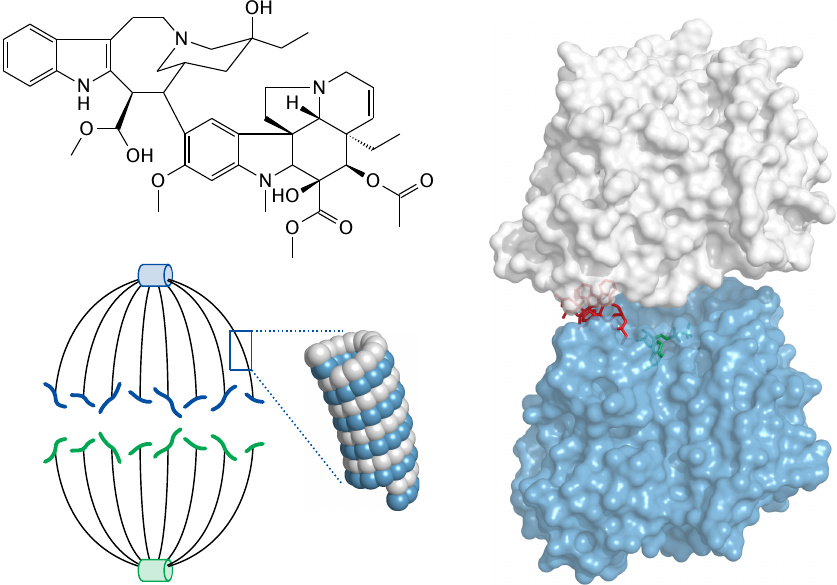
The mitotic spindle is the cytoskeletal apparatus that separates the chromosomes during the metaphase of mitosis. The spindle consists of microtubules, which are helical assemblies of tubulin αβ-heterodimers. Within the dimer, the drug vinblastine (red) binds between α-tubulin (white) and β-tubulin (blue). This distorts the geometry of the dimer and, by extension, that of the polymeric assembly. Tubulin structure rendered from 1z2b.pdb.
In addition to inhibiting mitosis, the disruption of tubulin polymerization also promotes apoptosis via activation of Bcl proteins. The molecular connection between these two events is only partially understood [126].
| 12.6.16 |
Alkylating anticancer drugs |
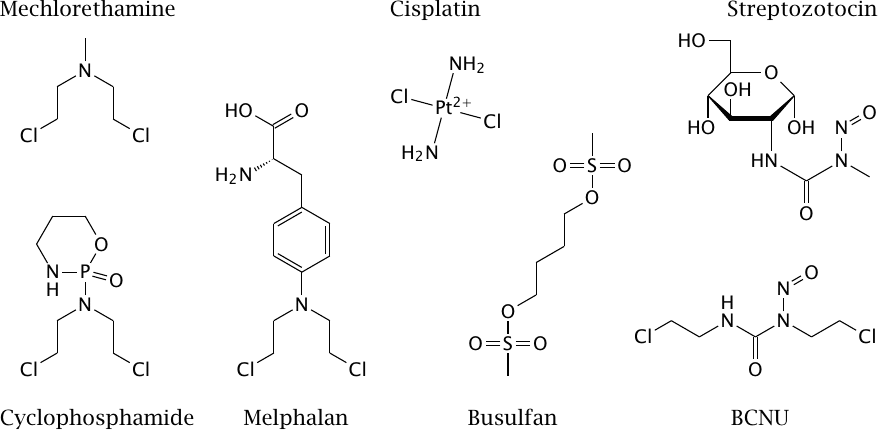
Alkylating drugs introduce covalent modifications into DNA and thus cause mutations and interference with replication. In contrast to the other types of cytotoxic drugs that we have covered so far, their effect is not limited to actively dividing cells but also affects resting cells. Since tumors typically contain a significant fraction of cells that are resting, alkylating drugs are part of most therapeutic drug combinations.
Two functional groups found in many alkylating drugs are the bis-chloroethyl-amine (N-mustard) group and the nitrosurea group. Mechlorethamine, cyclophosphamide, and melphalan all contain the N-mustard moiety, with its chloride leaving groups.106 Bis-chloroethyl-nitrosurea (BCNU, carmustine) and cisplatin have chloride leaving groups as well, whereas busulfan has methylsulfonate leaving groups. BCNU additionally contains the nitrosurea group, which is also found in the antibiotic streptozotocin.
| 12.6.17 |
Reaction of mechlorethamine with DNA |
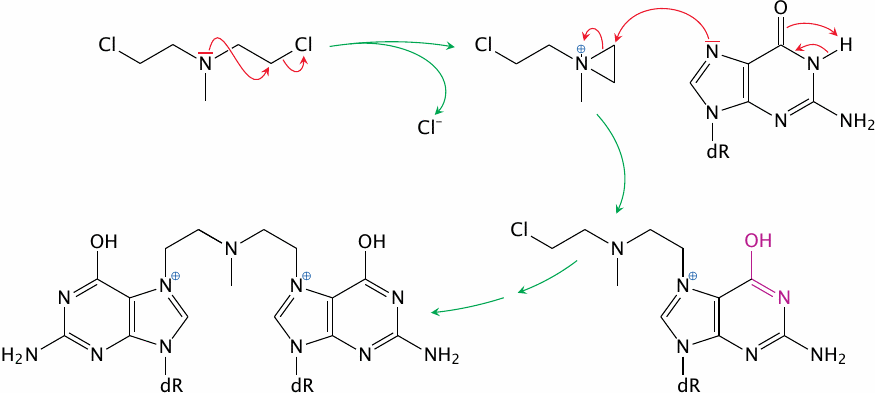
Mechlorethamine provides a straightforward example of DNA modification by the N-mustard drugs. The drug is activated by conversion of one chloroethyl group to the corresponding aziridine. The site in the DNA most likely to react with this aziridine is the N7 position of guanine. In the adduct, the aziridinium ring has been opened, which allows the next chloroethyl group to react. Successive reaction of two bases with the same drug molecule may crosslink the two DNA strands. Since such crosslinks are not amenable to excision repair, they form very effective and highly mutagenic lesions.
In the guanine adduct, the iminol tautomer is favored (highlighted in the first reaction step). This changes the base-pairing preference from cytosine to thymine and may cause mutations during DNA replication. In this manner, even non-crosslinked modified bases may give rise to mutation.
| 12.6.18 |
BCNU decay and adduct formation |
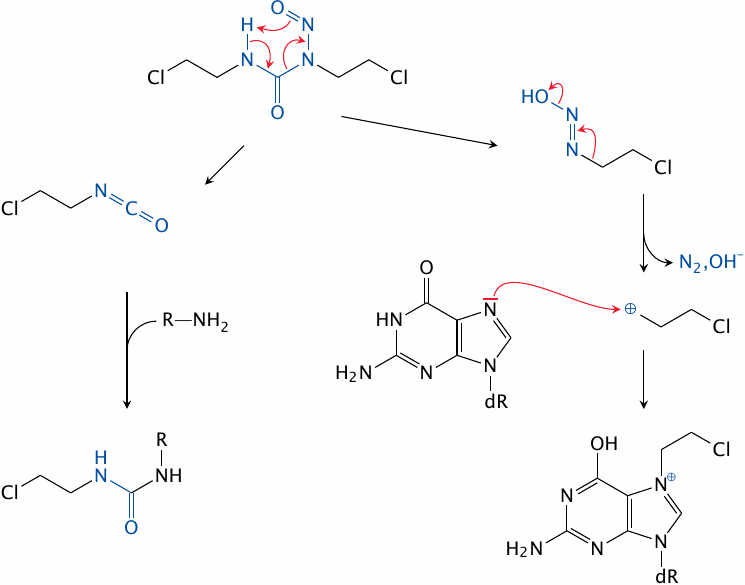
The nitrosourea moiety in BCNU decomposes spontaneously, yielding an isocyanate compound that reacts with primary amines (left) and a diazo compound that decays further into a carbocation, which in turn reacts with many nucleophiles, including bases in DNA (right). Each of the two BCNU fragments retains one chloroethyl moiety, which can engage in further reactions.
The antibiotic streptozotocin (slide 12.6.16) consists of a nitrosourea group attached to glucosamine. The sugar moiety facilitates its uptake by β cells in the pancreatic islets, which it selectively destroys. Streptozotocin is used therapeutically in β cell carcinomas, and experimentally in animals to induce diabetes mellitus.
| 12.6.19 |
Reaction of daunorubicin with DNA |
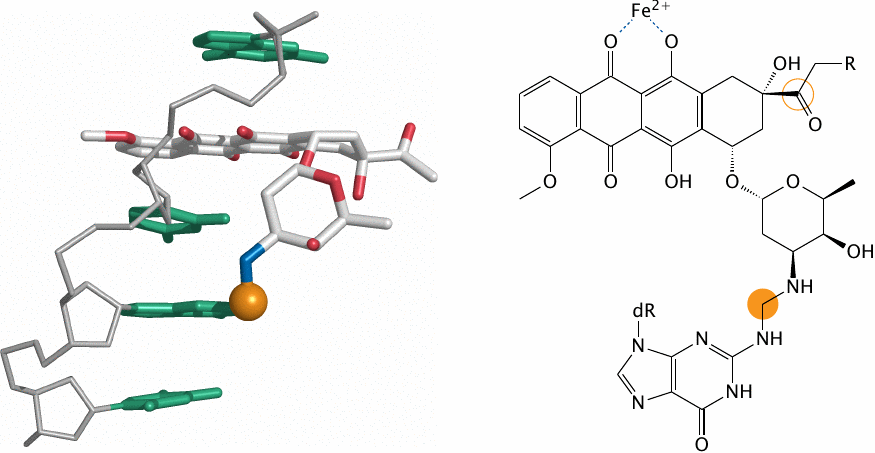
Daunorubicin (R=H) and doxorubicin (R=OH) are antibiotics produced by a soil bacterium (Streptomyces peucetius). Mediated by their planar polycyclic rings, these molecules intercalate between the stacked bases of DNA double strands (but only one DNA strand is shown here; structure rendered from 2d34.pdb.). The amino group on the daunosamine sugar moiety of the drug then becomes linked by a methylene bridge to the N2 amino group of a guanine base (highlighted).
The carbon that forms this bridge is derived from formaldehyde, which arises from cellular precursors or, with doxorubicin, from the hydroxyacetyl side chain on the drug itself (also highlighted) through a series of redox reactions that occur between glutathione, iron and the drug’s quinoid rings.
Another antibiotic that has been (but no longer is) used in cancer chemotherapy is calicheamicin (see slide 14.5.2).