| 13 |
Ribonucleic acids as drugs and drug targets |
| 13.1 |
Introduction |
As we have seen in the preceding chapters, most drugs target proteins, but some drugs act on ribonucleic acids (RNAs) instead. Among these, the currently most widely used ones are antibiotics that inhibit the bacterial ribosome (see slide 11.5). However, as more has been learned about the diverse and crucial roles of RNA in eukaryotic cell biology, novel RNA drug targets have emerged and become a focus of ongoing drug development efforts.
In this chapter, we will first have a closer look at some of the antibiotics that interact with the RNA of the bacterial ribosome, and then turn to some of the experimental strategies that target RNA molecules in human cells.
| 13.2 |
Antibiotics that inhibit the bacterial ribosome |
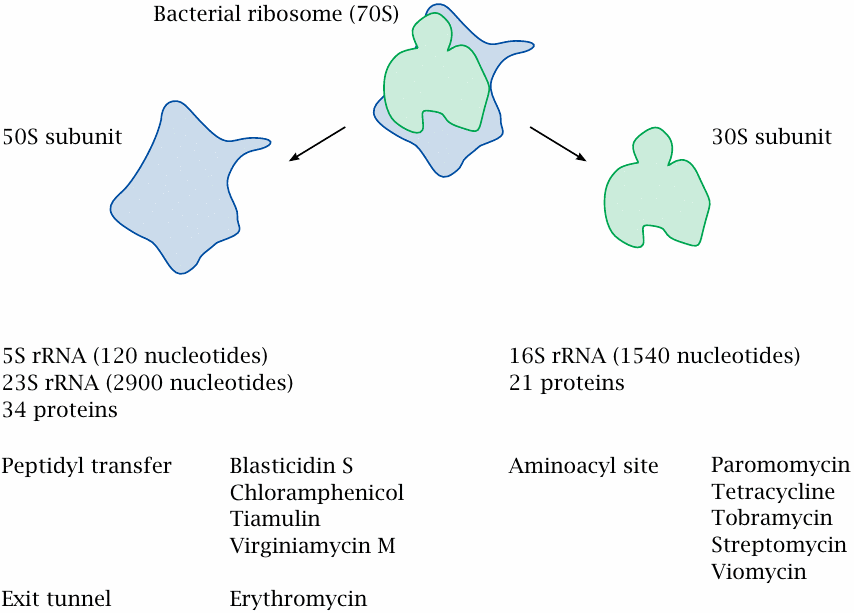
The bacterial ribosome consists of two subunits that can be separated under mild conditions. Different classes of antibiotics bind to either of these, and in the case of the larger subunit to distinct functional sites within. While ribosomes consist of both RNA and protein molecules, structural studies have determined that most of the interactions with antibiotics involve the ribosomal RNA rather than the ribosomal proteins.
| 13.2.1 |
Some aminoglycoside antibiotics |
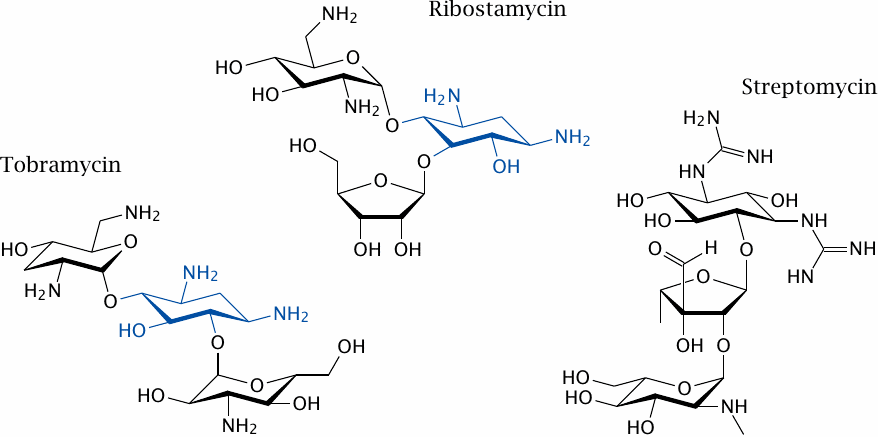
Aminoglycoside antibiotics are produced by Streptomyces species and related soil bacteria. As the name suggests, they are oligosaccharides substituted with primary or secondary amines and related functional groups. These antibiotics bind to the aminoacyl acceptor site in the small subunit of the bacterial ribosome.
Among the antibiotics shown here, ribostamycin is a “typical” aminoglycoside that belongs to the 4,5-disubstituted deoxystreptamine group, and tobramycin a typical aminoglycoside with 4,6-disubstituted deoxystreptamine (the deoxystreptamine moiety is shown in blue). Streptomycin does not contain a deoxystreptamine group and thus is an “atypical” aminoglycoside.
Tobramycin is clinically important in the treatment of infections due to Gram-negative bacteria and in particular Pseudomonas aeruginosa, against which it counts among the most effective antibiotics. Streptomycin is used in combination with other antibiotics in treating tuberculosis.
| 13.2.2 |
Paromomycin in the ribosomal aminoacyl acceptor site |
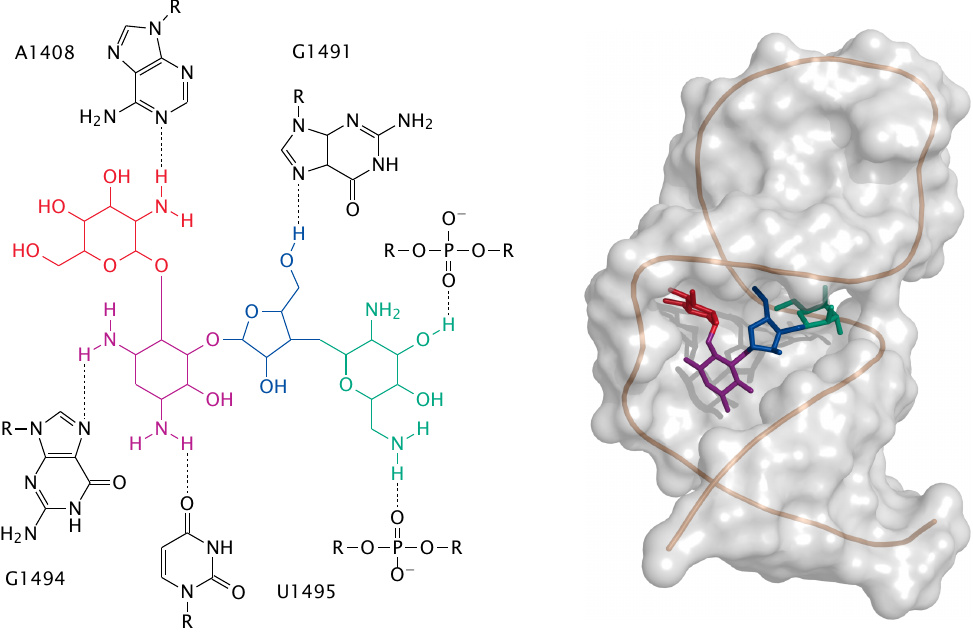
Paromomycin is another aminoglycoside antibiotic. This three-dimensional structure of the RNA-paromomycin complex was determined by NMR spectroscopy, using a short model RNA that mimics the crucial double-stranded section of the (much larger) ribosomal RNA [127]. Rendered from 1pbr.pdb.
The “flattened” structure on the left shows the network of hydrogen bonds between the drug and the nucleotides of the ribosomal RNA. It can be seen that bonds involve both the bases and the backbone phosphates of the RNA.
| 13.2.3 |
Inactivation of kanamycin by resistance enzymes |
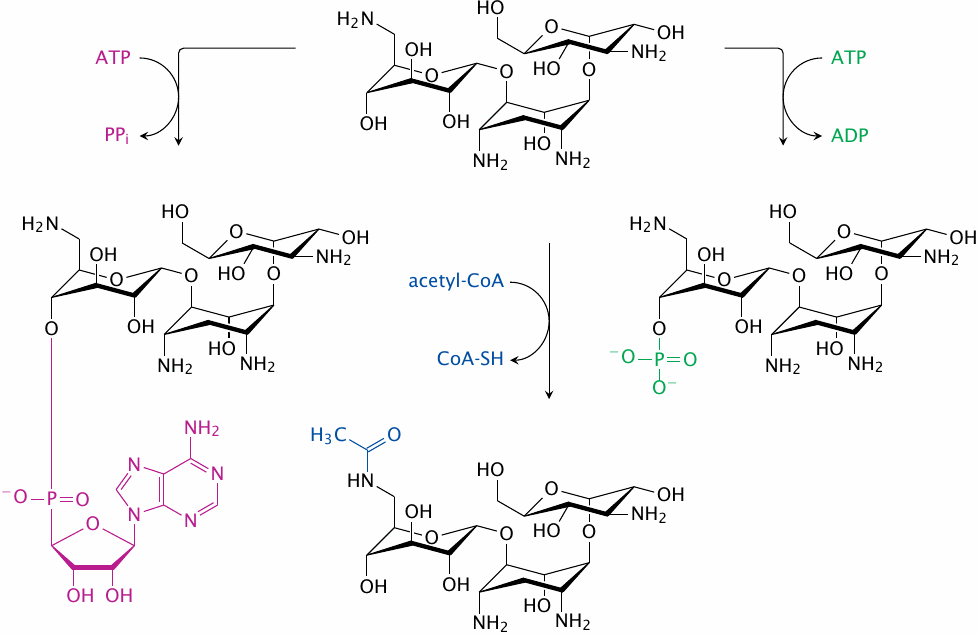
Aminoglycosides will be rendered inactive by covalent modification of the amino and hydroxyl groups that form hydrogen bonds with the bacterial RNA. Enzymes that perform such covalent modifications mediate bacterial resistance to these antibiotics.
This slide illustrates the reactions catalyzed by several resistance enzymes that inactivate kanamycin A, which is yet another aminoglycoside. Adenylylation (purple) is catalyzed by aminoglycoside O-adenylyltransferases, acetylation (blue) by amino-glycoside N-acetyltransferases, and phosphorylation (green) by aminoglycoside O-phosphotransferases. The semisynthetic kanamycin derivative amikacin has been functionalized on an amino group that is not essential for binding to the ribosome. This modification protects the drug from some resistance enzymes. Amikacin is valuable in infections with Gram-negative bacteria that are difficult to treat otherwise.
| 13.2.4 |
Interactions of chloramphenicol with RNA in the peptidyl transferase site of the ribosome |
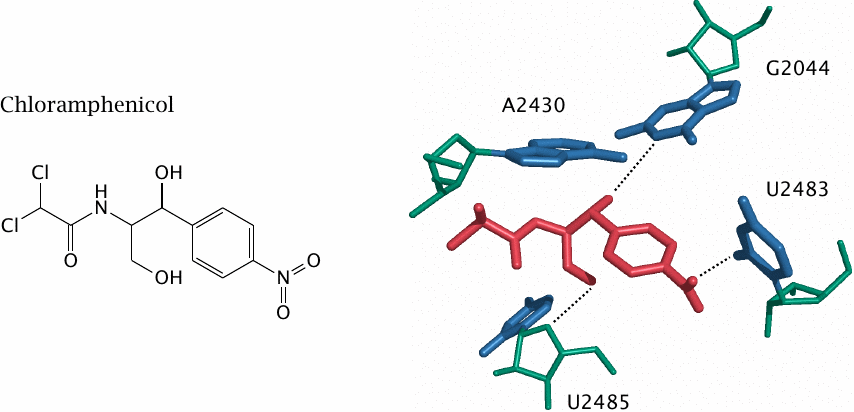
Chloramphenicol is a small, fairly lipophilic molecule with a broad spectrum of antibacterial activity and very favorable pharmacokinetic properties, but it is almost never used in developed countries because of allergic bone marrow toxicity.107 The drug binds within the peptidyltransferase site of the ribosome (see slide 11.5.1), which is part of the large subunit of the ribosome.
Like paromomycin, chloramphenicol interacts exclusively with the ribosomal RNA; this interaction involves both of its hydroxyl groups. The acetylation of one hydroxyl group by chloramphenicol acetyltransferase is an important resistance mechanism.
| 13.3 |
Novel RNA drug targets |
The antibiotics discussed above show that ribosomal RNA makes a perfectly fine drug target, which suggests that RNA targets other than ribosomes should also be amenable to small drug molecules. This paradigm is still at the experimental stage.
| 13.3.1 |
The RNA component of human telomerase |
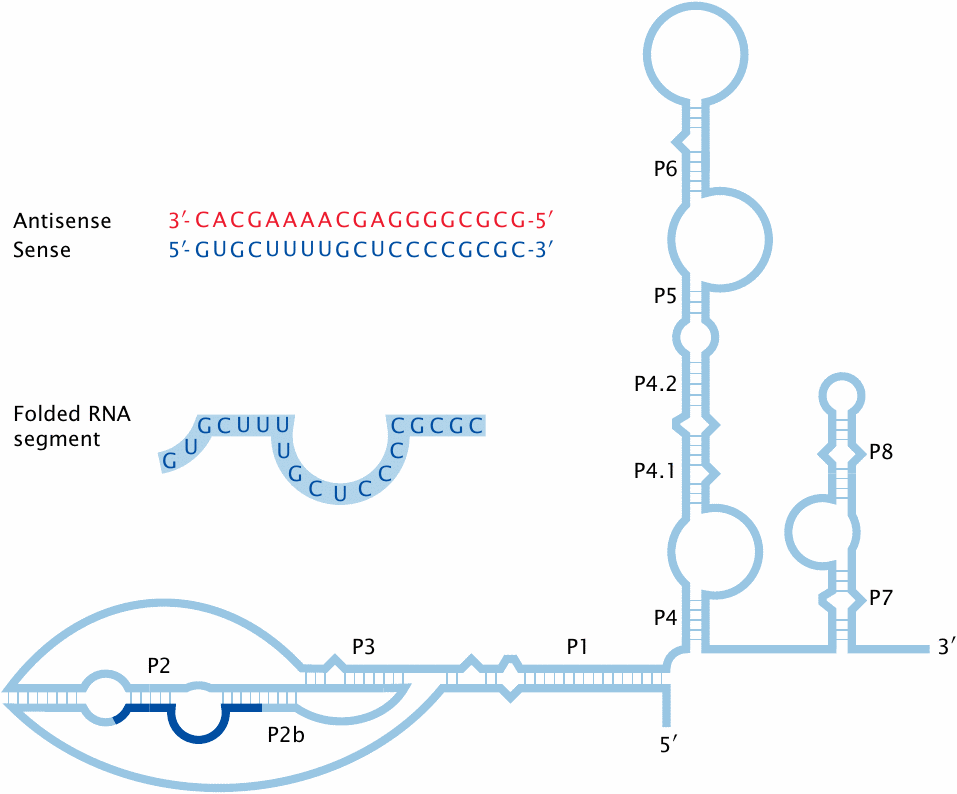
Telomerase is an enzyme that synthesizes repetitive DNA segments and attaches them to the telomeres, that is, the free ends of the chromosomes. Telomeres get shortened during each mitotic cell division, and if not renewed by telomerase, cumulative shortening during repeated cell divisions will ultimately wear them down entirely, putting an end to cell proliferation.
As one might guess from the foregoing, telomerase is often overexpressed in tumor cells, contributing to their ability to proliferate indefinitely. This has prompted research into telomerase inhibitors as anticancer agents. The enzyme consists of both protein and RNA moieties. The RNA component, which serves as a template for the polymerization of DNA, is essential for the enzyme’s function, and therefore suitable in principle as a drug target. One class of inhibitory molecules are antisense oligonucleotides, which hybridize to some strategic part of the target RNA and thereby disrupt its function.
The picture shows the secondary structure of the human telomerase RNA component. A possible binding site for inhibitory antisense oligonucleotides is highlighted. The antisense nucleotide shown will base-pair with all bases in the target sequence, including those that are not base-paired within the native RNA molecule. This will maximize the affinity of the antisense molecule for the target.
| 13.3.2 |
Unusual nucleotide linkages in synthetic oligonucleotides |
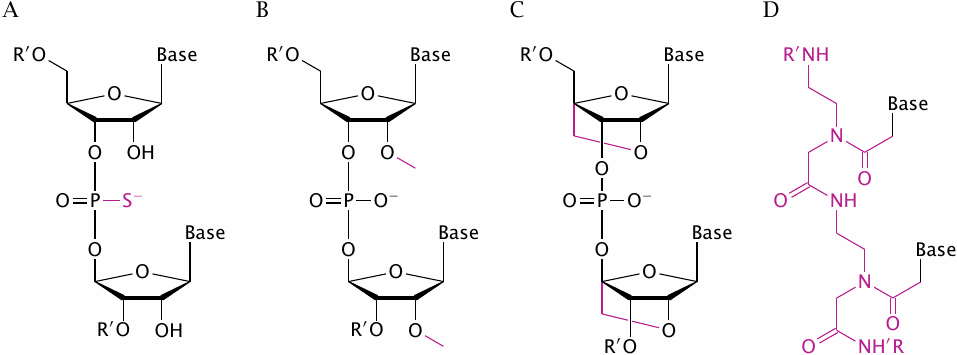
The above example of telomerase illustrates the conceptual elegance and simplicity of the RNA antisense approach: Affinity and specificity of an oligonucleotide for a given target are much more predictable than is the case with small organic molecules that bind to proteins. Nevertheless, antisense oligonucleotides have found very few applications in clinical practice. Why is this?
Compared to conventional drug molecules, antisense oligonucleotides are rather large—they must contain approximately 15–20 bases to achieve sufficient specificity for a unique target sequence—as well as polar, which is due to the phosphodiester bonds in the backbone. These phosphodiester bonds are also subject to rapid degradation by RNAse or DNAse enzymes. Therefore, overall, oligonucleotides have very unfavorable pharmacokinetic properties.
To increase stability in vivo, synthetic oligonucleotide analogues with modified backbone structures have been developed. The picture shows some examples; the structural deviations from regular RNA backbone structure are highlighted.
Phosphorothioates (A), 2′-O-methyl RNA (B) and “locked nucleic acids” (LNA; C) have modified backbones that render them resistant to degradation by nucleases. In “peptide nucleic acids” (PNA, D), the sugar-phosphate backbone is replaced entirely with a polyamide structure that confers not only resistance to cleavage but also reduces polarity.
| 13.3.3 |
The HIV transactivation-responsive region (TAR) |
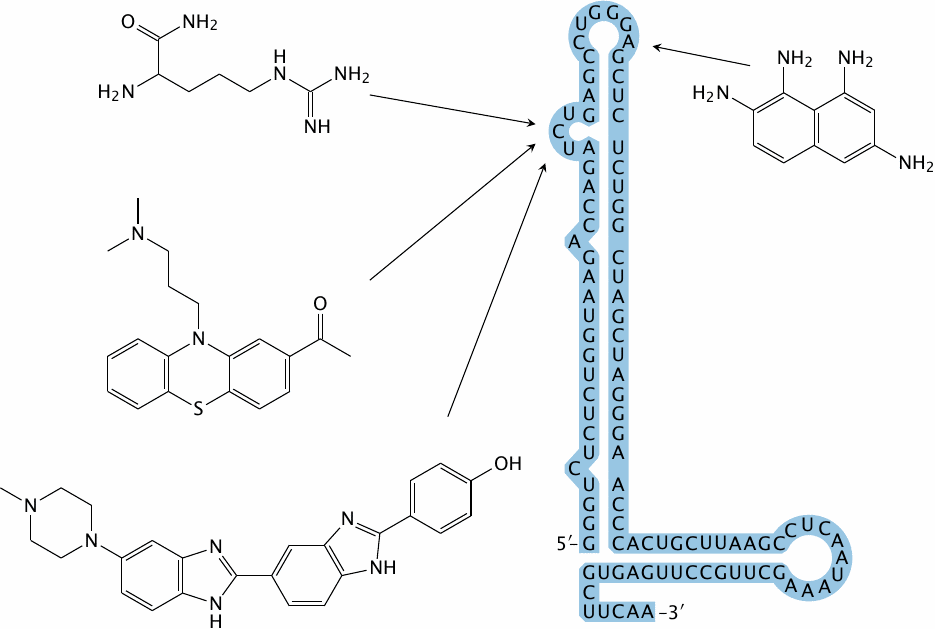
The transactivation-responsive region (TAR) is an RNA motif that occurs at the 5′-end of human immunodeficiency virus (HIV) RNA transcripts. On a nascent transcript, this region forms a characteristic stem-loop structure that is recognized by the viral tat protein, which then recruits cellular proteins to assist with the completion of transcription. Disrupting the interaction between TAR and tat might be a viable strategy for inhibiting HIV multiplication.
The figure shows the secondary structure of TAR, as well as several small molecules that have been screened from a library for binding to this RNA [128]. The arrows indicate the approximate binding sites within the TAR.
| 13.3.4 |
Blocking premature translational termination with PTC124 (ataluren) |
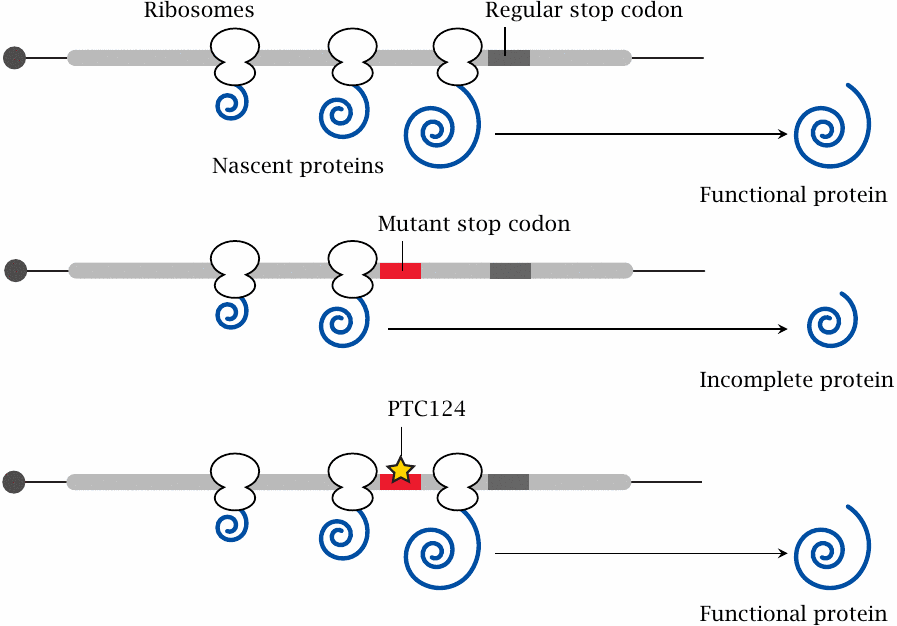
Many genetic diseases are caused by point mutations that inactivate some important enzyme or other proteins. Very often, such inactivating mutations are diverse, affecting different positions within the reading frame of the protein in question, and either causing the replacement of a functionally crucial amino acid residue, or creating a premature stop codon. In the latter case, it may be possible to restore expression of a functional protein by promoting translational miscoding, that is, incorporation of some amino acid or other vis-a-vis the mutant stop codon, which then enables the ribosome to keep translating right through to the regular stop codon.108
An experimental drug that promotes translational suppression of premature stop codons is PTC124 [129], also named ataluren. Its biochemical mode of action is not known exactly; however, it is noteworthy that several aminoglycoside antibiotics have similar effects, and some are also being investigated for similar therapeutic uses [130].
| 13.3.5 |
Ataluren in cystic fibrosis |
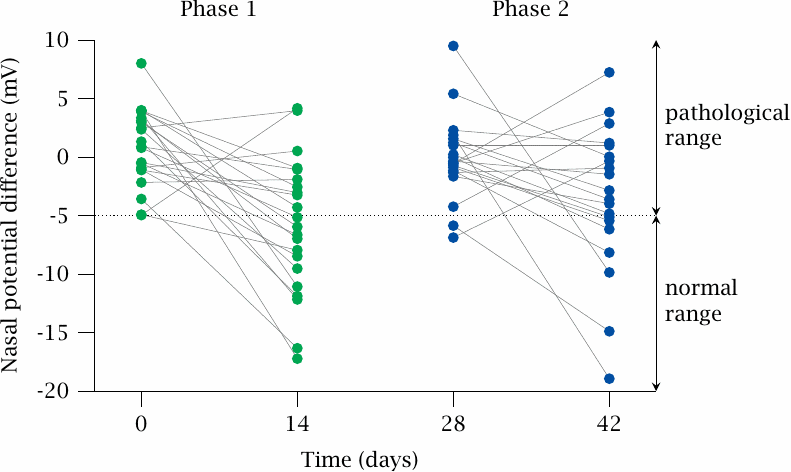
The effectiveness of ataluren in various genetic diseases has been tested in clinical pilot studies. This slide illustrates one such study, which was performed on cystic fibrosis patients. This disease is caused by a deficiency in a membrane transport protein that exports chloride ions from cells, which impedes the secretion of fluid in all kinds of exocrine glands. Multiple organs are functionally impaired, but the most serious consequences arise from the build-up of viscous mucus in the lungs, which facilitates chronic bacterial infections and ultimately leads to organ destruction.
In a sizable minority of all CF patients, the gene defect consists in a premature stop codon, and in this group translational antitermination is a plausible therapeutic strategy. The data illustrate the response to the drug among such patients, during two successive phases of treatment. The effect of the drug is measured as the potential that exists across the nasal mucous membrane; this potential is affected by the capacity of the epithelial cells to export chloride. Lines connect measurements on individual patients before and after each course of treatment. Figure prepared from original data in [131].
| 13.3.6 |
RNA interference |
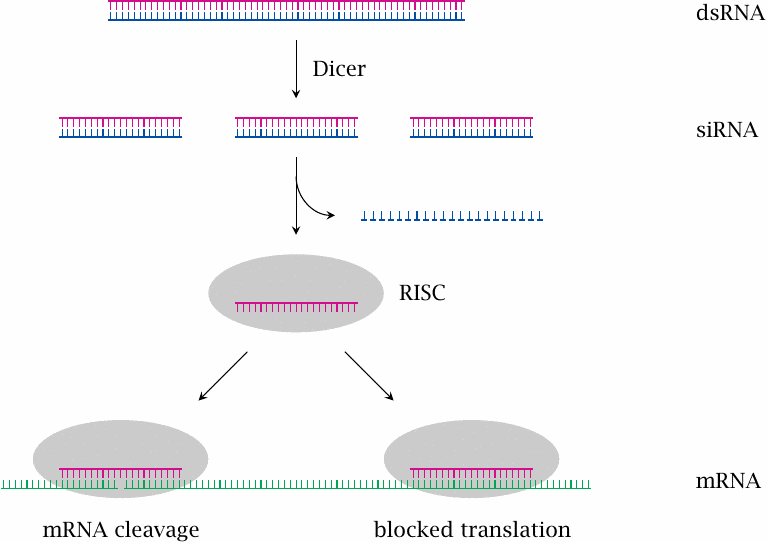
In RNA interference, the presence of double-stranded RNA (dsRNA) suppresses the translation of a corresponding single-stranded messenger-RNA. In this process, the dsRNA is cleaved into small fragments by an enzyme known as dicer. The double-stranded fragments—referred to as silencing RNA, or siRNA—are picked up by RNA-Induced Silencing Complex (RISC), which contains multiple protein subunits and catalytic activities. This complex degrades one strand of the siRNA and uses the other to bind to the complementary mRNA, which it inactivates either simply through binding as such, or through subsequent cleavage.
RNA interference is important as a non-specific immune mechanism against the proliferation of RNA viruses, which must go through a dsRNA stage during genome replication and transcription.109 However, RNA interference is not limited to viral RNA as a target, and synthetic siRNA is now often used in cell culture experiments to inhibit the expression of specific genes, for example to validate the corresponding protein as a drug target. Application of siRNA in pharmacotherapy is hampered by the same problems that also afflict antisense oligonucleotides.
| 13.3.7 |
The SELEX process for generating RNA aptamers |
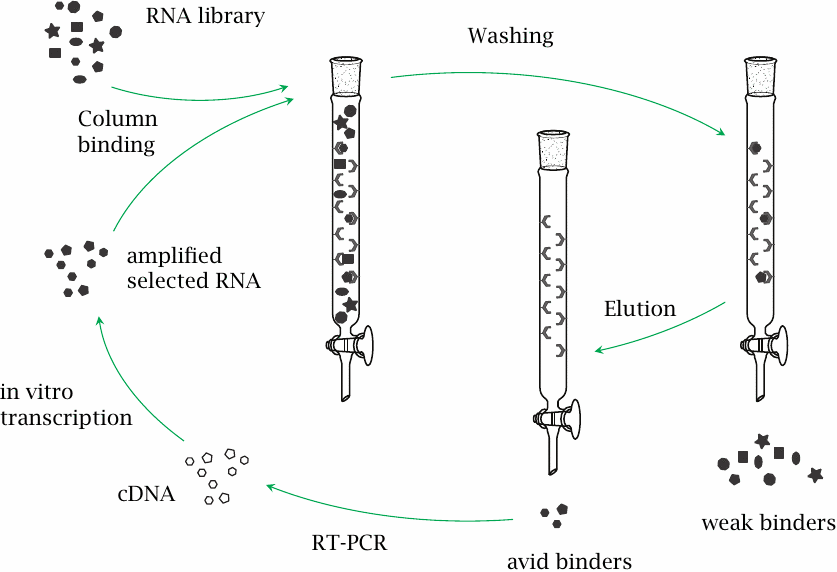
RNA aptamers are artificial RNA molecules with affinity for a given ligand of interest. They are of potential interest for therapeutic use in a manner similar to therapeutic antibodies, although pharmacokinetic limitations again restrict such uses in practice.
RNA (and similarly, DNA) aptamers can be obtained through an ingenious combination of molecular biology and conventional affinity chromatography that is referred to as systematic evolution of ligands by exponential enrichment, or SELEX for short. In such an experiment, the ligand of interest is bound to a solid phase within a chromatography column. A library of random RNA sequences is incubated with the immobilized ligand, whereafter RNAs that remained unbound are washed away. Ligand-bound RNAs are eluted and amplified through RT-PCR and in vitro transcription, and the process repeated using more stringent conditions for binding and washing. PCR conditions can be chosen so as to encourage mutations, which may further enhance the affinity of the selected aptamers.