| 6 |
Pharmacology of cell excitation |
| 6.1 |
Overview |
Nerve cells, muscle cells and several other cell types are excitable, that is, they are activated by variations of their membrane potentials. Many of the macromolecules that control cell excitation are important drug targets.
| 6.1.1 |
Clinical applications of drugs that influence excitable cell function |
- blockade of nerve conduction for local anesthesia
- reduction of nerve cell excitability in the brain in epilepsy
- stabilization of mood in the treatment of bipolar disorder
- reduction of vascular smooth muscle tone to reduce blood pressure
- suppression of aberrant excitation in cardiac arrhythmia
This list is not complete, but it suffices to illustrate that the applications are important and diverse. In order to understand how such drugs work, we must first understand cell excitation itself.
| 6.1.2 |
The nature of cell excitation |
- all cells have an electrical potential across the cytoplasmic membrane, such that the cell interior is electrically negative relative to the outside (~−70 mV)
- in non-excitable cells, this membrane potential is stable; in excitable cells, it forms the resting potential
- cell excitation consists in transient reversals of the membrane potential, called action potentials, which spread rapidly across the entire cell membrane
- action potentials are spontaneously generated by some cells and transmitted between cells through chemical or electrical synapses
By definition, a negative membrane potential means that the cell interior is negative relative to the extracellular space. The negative resting potential is maintained by potassium leak channels in the cytoplasmic membrane; we will consider below how this works.
| 6.1.3 |
Neurons and synapses |
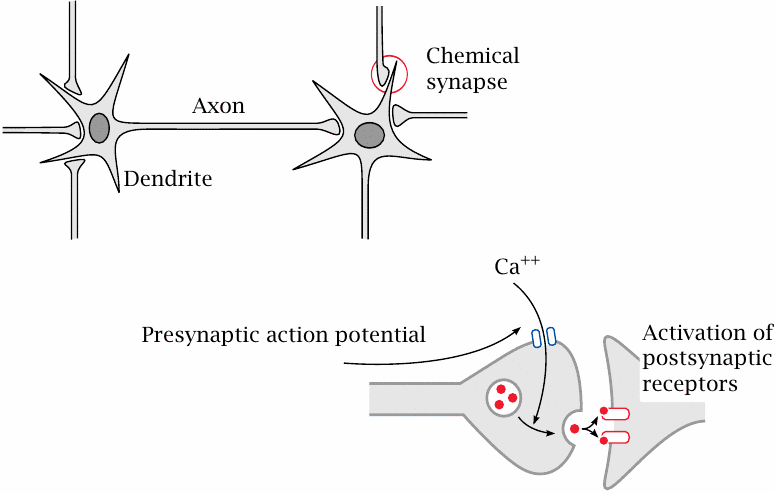
Many drugs that influence cell excitation act on nerve cells. Nerve cells, or neurons, communicate with one another through chemical synapses. A nerve cell receives input from other nerve cells upstream through synapses on its dendrite. When stimulated sufficiently by activity of these synapses, it will generate an action potential. The action potential travels down the dendrite toward the cell body, then down the axon. When it reaches a presynaptic terminal, the action potential causes an influx of Ca++, which in turn triggers the release of neurotransmitter into the synaptic cleft. Activation of postsynaptic receptors then transmits the signal to the next nerve cell.
A given neuron receives input from its afferent synapses, and it sends its output to its efferent synapses.
| 6.1.4 |
Some nerve cells have huge dendrites and axons |
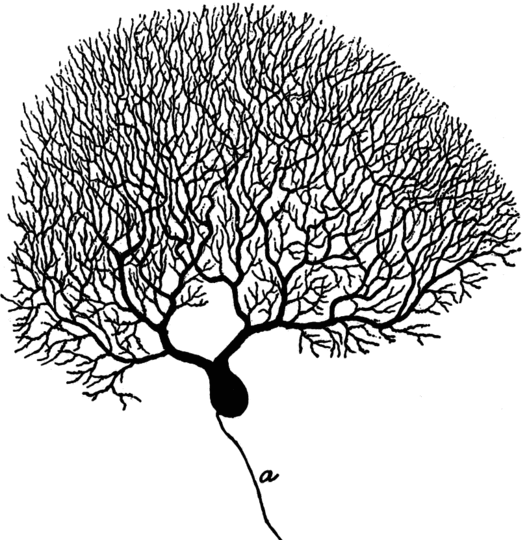
This picture (from [42]) shows a Purkinje cell from the cortex of the cerebellum. It has a large branched dendrite,33 through which it receives input from thousands of other nerve cells.
The axon of the Purkinje cell has far fewer branches than its dendrite (and they would all be below the lower edge of this picture). However, in some other types of nerve cells, the axon may be similarly strongly branched. For example, a single α-motoneuron may control the activity of several hundred skeletal muscle fibers; its axon must branch out and form synapses with each of these cells.
| 6.1.5 |
Other types of excitable cells |
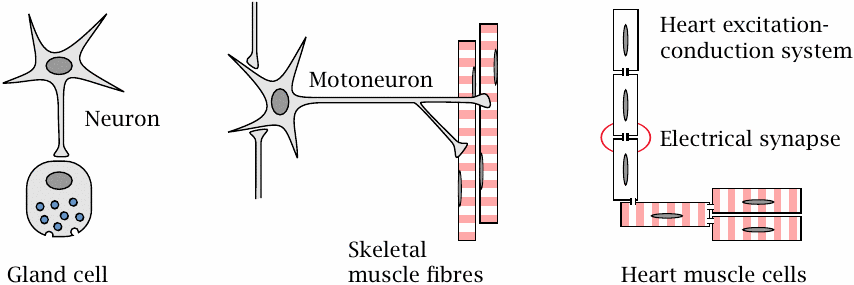
In addition to nerve cells, several other types of cells are also excitable and may contain clinically relevant drug targets. Some of these cells receive their input from nerve cells. Adrenal gland cells release norepinephrine and epinephrine when stimulated by nerve cells of the autonomic nervous system. Skeletal muscle cells contract when stimulated by α-motoneurons; vascular smooth muscle cells contract in response to stimulation by both nerve cells and circulating hormones.
The heart, while also subject to modulatory input from the autonomic nervous system, can function entirely independently.34 It possesses two types of excitable cells. Those within the excitation-conduction system spontaneously produce the rhythm, and they then transmit it to the regular muscle cells, which we will here refer to as the “worker” cells. Communication occurs through electrical synapses, in which ions can flow directly from cell to cell via cytoplasm bridges. These direct electrical connections between adjacent cells are created by so-called gap junctions.
| 6.2 |
The physical basis of cell excitation |
Now comes the scary part—the one we all thought we had successfully dodged when choosing bioscience over real science.
| 6.2.1 |
The two driving forces that generate diffusion potentials across membranes |
| \(\Delta E\) | = | \(\frac{\text{RT}}{\text{zF}}\:\text{ln}\frac{[\text{cation}]_{\text{left}}}{[\text{cation}]_{\text{right}}}\) |
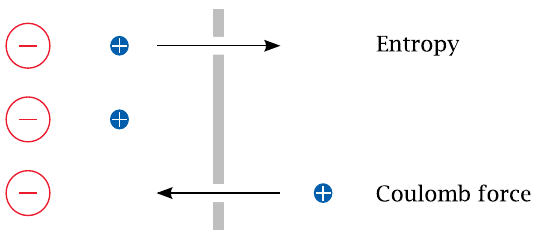
A diffusion potential arises if (1) the membrane is selectively permeable for some specific ion but not its counterion, and (2) there is a concentration gradient for the permeant ion. Under these conditions, entropy will induce the permeant ion to move downhill its concentration gradient, until the ensuing charge imbalance creates an equally strong Coulomb counterforce. This counterforce is reached at the equilibrium potential, E0, which is determined by the Nernst equation.35
| 6.2.2 |
Equilibrium potentials for the major salt ions |
| Ion | Cytosolic | Extracellular | E0 at 37∘C |
| K+ | 150 mM | 6 mM | – 86 mV |
| Na+ | 15 mM | 150 mM | + 62 mV |
| Ca++ | 100 nM | 1.2 mM | + 126 mV |
| Cl− | 9 mM | 150 mM | – 70 mV |
This table lists the major cations and anions that control the membrane potential and their intra- and extracellular concentrations. For each ion, the equilibrium potential was calculated using the Nernst equation.
As you can see, the equilibrium potentials are all different; those of K+ and Cl− are negative, while the ones for Na+ and Ca++ are positive. This raises the question: How do these individual, different equilibrium potentials interact to shape the actual membrane potential? To understand this, we need to consider another parameter, namely the permeability of the membrane for each ion.
Pure phospholipid membranes are essentially impermeable for salt ions; therefore, in cell membranes, ion permeabilities are determined by specific transport proteins, the ion channels. Ion channels that open and close and thereby cause ion permeabilities to change are at the heart of cell excitation.
| 6.2.3 |
Specific channels control ion permeabilities |
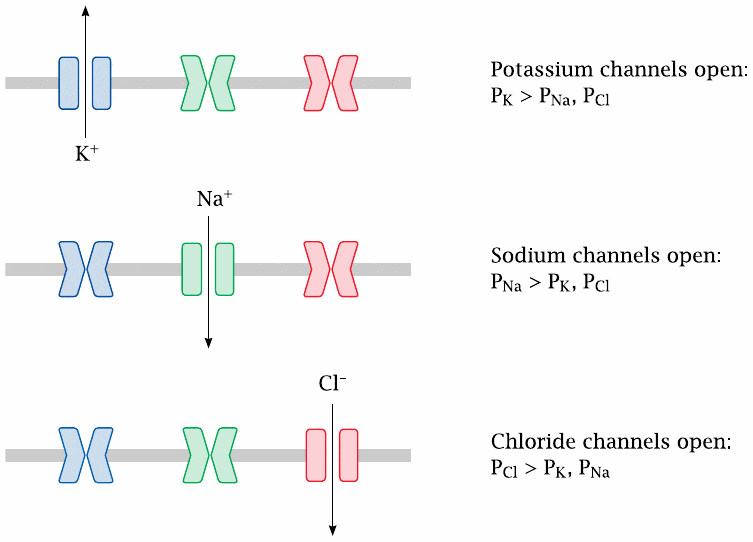
Some ion channels are continuously open, but most open transiently and then close again. Within this latter group, ligand-gated channels open in response to binding of a chemical agonist, while voltage-gated channels respond to changes in the membrane potential. Some channels may respond to both ligands and voltages. Yet others respond to changes in temperature or membrane distension; such channels are significant in sensing heat and pressure.
Many channels are very specific for individual ion species, as shown in this illustration; this applies to the major voltage-gated channels that control the action potential. Other channels have broader selectivity; for example, the nicotinic acetylcholine receptor (slide 6.10.1) is permeable for all of the major cations (Na+, K+ and Ca++).
In this slide, P represents the ion-specific membrane permeabilities that are explained in the next slide.
| 6.2.4 |
Diffusion potentials with multiple ions: the Goldman equation |
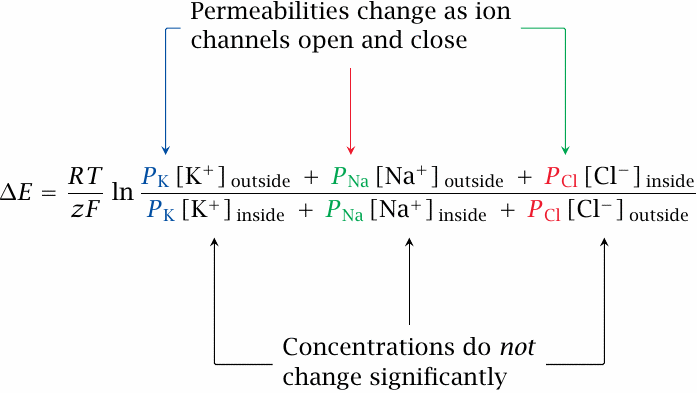
The diffusion potential that prevails across a membrane with significant permeabilities for several ion species is governed by the Goldman equation, shown here for a combination of Na+, K+ and Cl−. In addition to the terms known from the Nernst equation, it introduces the permeability P, a scaling factor that weights each ion for its rate of diffusion across the membrane, relative to that of the other ions.
If channels open for a specific ion, its permeability goes up. This increases the weight of this ion in the Goldman equation, and thus pulls the overall membrane potential toward its own (Nernst) equilibrium potential. In resting cells, the potential is close to the K+ equilibrium potential, because the always-open K+ leak channels give K+ a greater permeability than all other ions. However, all that is needed to flip the membrane potential from its negative-inside resting state to a positive-inside value is a forceful opening of sodium or calcium channels. On the other hand, opening of chloride channels or of additional potassium channels will drive the membrane potential back down toward the resting state or below.
As stated above, such transient reversals of the membrane potential are referred to as action potentials. In nerve and skeletal muscle cells, an action potential lasts only 1–3 milliseconds, and the absolute number of ions that cross the membrane during this short time interval is very low. With Na+, K+ and Cl−, it is too low to measurably change the ion concentration on either side of the membrane.
The one important exception from this rule is calcium. Its intracellular concentration is so low (see slide 6.2.2) that the small number of ions flowing across open channels still causes a significant relative increase. The increased intracellular calcium level will cause activation of calmodulin and other calcium-binding proteins. Therefore, the opening of calcium channels constitutes both a physical and a biochemical signal. Calcium channels are important in nerve cells, and they are particularly prominent in heart and smooth muscle cells, where they replace sodium channels as the primary drivers of membrane potential reversal, or depolarization.
| 6.3 |
Structure and function of voltage-gated cation channels |
Voltage-gated cation channels are crucial in triggering, sustaining and propagating action potentials. The channels for K+, Na+ and Ca++ are homologous and functionally similar. We will look at the structure of voltage-gated K+ channels as an example.
| 6.3.1 |
Structure of a voltage-gated K+ channel |
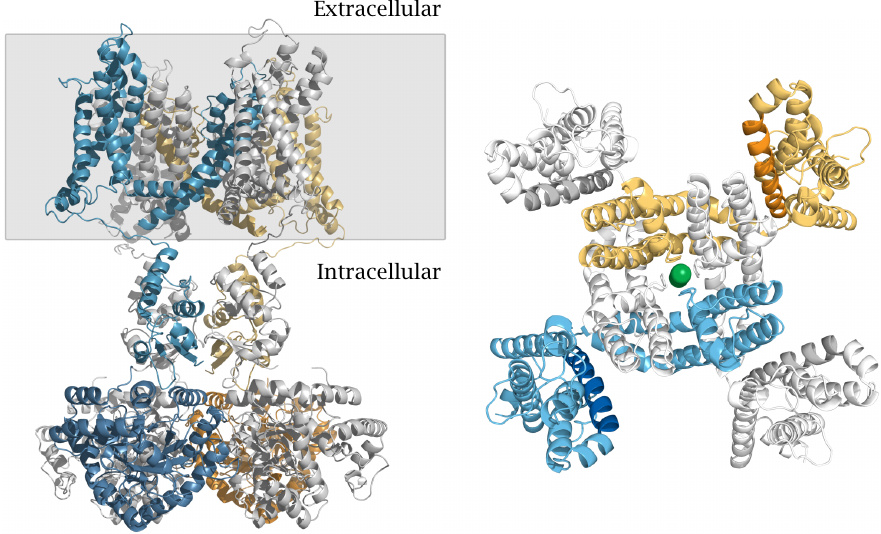
The picture on the left shows a side view. The gray rectangle delineates the membrane-embedded portion. A large part of the channel protein protrudes into the cytosol. The channel is composed of four identical subunits; one is shown in blue, the one opposite to it in yellow, and the other two in light gray. The conformationally flexible N-terminal inactivation domain that is located in the cytosolic portion (see below) is missing from this structure.
The panel on the right shows a view from the extracellular side onto the membrane-embedded domain of the channel. This domain consists of an inner layer, which contains the selectivity filter, and an outer layer that mediates voltage-dependent channel opening. In each of the four subunits, an arginine-rich helix that forms the voltage sensor is rendered in a darker shade. A potassium ion inside the central channel pore is shown as a green ball. While most of the channel protein is α-helical, the selectivity filter that enwraps the potassium ion has β-structure. Structure rendered from 2r9r.pdb.
| 6.3.2 |
Structure of the K+ selectivity filter |
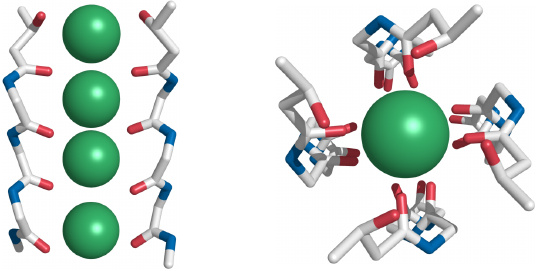
Side view (left) and top view (right) of the K+ selectivity filter that is in the center of the membrane-embedded portion. Backbone oxygen atoms tightly enclose the dehydrated K+ ions. Each of the four subunits contributes one backbone segment, but for clarity only two segments are shown in the side view.
Similar selectivity filters are also found in other types of K+ channels, for example the leak channels that maintain the resting potential, as well as the KATP channels (slide 6.6.1). The selectivity filters found in sodium and calcium channels work according to the same principle, although they differ in diameter and architectural detail. In each case, the filter fits the ion like a glove. Ions that are too large are simply excluded; those that are too small are rejected because they cannot bind avidly to the filter, and therefore cannot compensate the energetic cost of their dehydration.
| 6.3.3 |
Voltage-gated sodium channels sustain and spread the action potential |
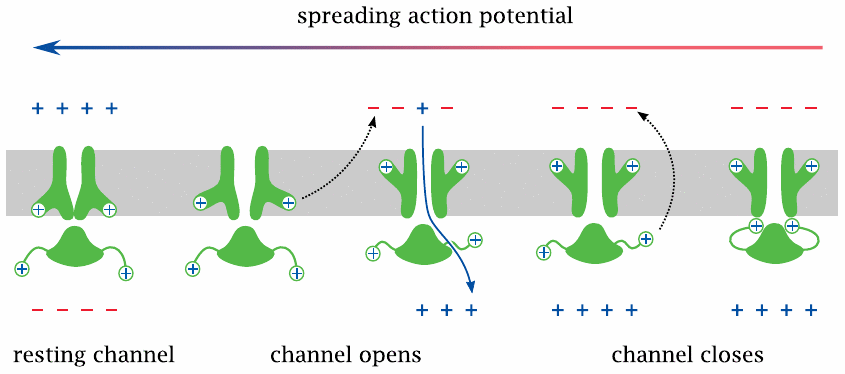
On nerve and skeletal muscle cells, the action potential is carried and sustained mostly by voltage-gated Na+ (NaV) channels. As stated above, all voltage-gated channels have the same general structure as the KV channel, so we will now see how those structural features operate in practice.
When the membrane potential in the vicinity of a sodium channel is reversed by a spreading action potential, the membrane-embedded voltage sensor moves outward, which decompresses the inner layer and opens the channel. This allows Na+ to enter the cell, enlarging the depolarized membrane area, causing more NaV channel molecules to open and the action potential to propagate further.
After the channel has been open for a short time,36 a separate inactivation gate, which is flexibly attached to the cytosolic portion of the channel and also carries a positive net charge—and which, as stated, is missing from the KV channel structure shown in slide 6.3.1—responds to the same electric field reversal and moves to plug the channel. The channel is now inactivated, in spite of the continued depolarized state of the membrane. Before the channel becomes ready for opening again, both gates must revert to their initial conformations, which happens only after the membrane has been repolarized.37
While the explanation given here is mostly quite accurate, we must add one caveat: the membrane potential does not have to be fully reversed in order to induce channel opening; instead, the channels begin to open once the membrane has been partially depolarized to approximately –50 mV. The threshold potentials or “firing levels” of different channels do vary, but they are at negative values for all voltage-gated channels I’m aware of.
| 6.3.4 |
Voltage-gated potassium channels extinguish the action potential |
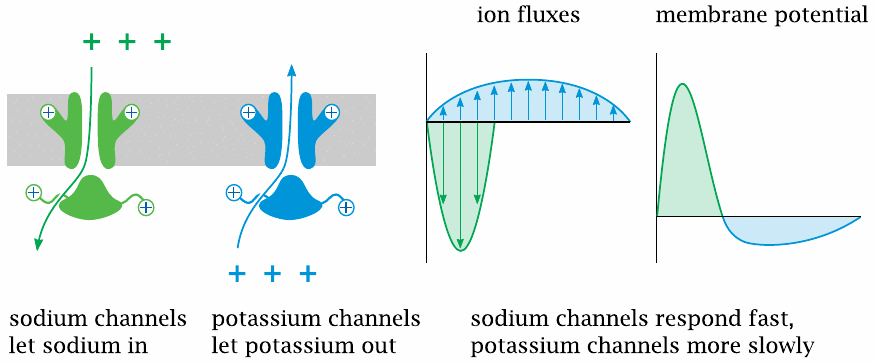
This slide illustrates the interplay of NaV and KV channels in shaping the action potential. Both channels open in response to depolarization. Because of the opposite orientations of the two concentration gradients, this causes Na+ influx but K+ efflux.
The NaV channels open and inactivate faster and therefore dominate the early (depolarization) phase of the action potential. The slower response of the KV channels dominates the later phase and causes a transient hyperpolarization of the membrane.
As long as the composition of the channel population remains the same, the shape and amplitude of each action potential will also be the same.38 Hence, information cannot be encoded in the properties of individual action potentials; instead, it is encoded in their frequency.
| 6.3.5 |
Voltage-gated channels and action potentials in the heart |
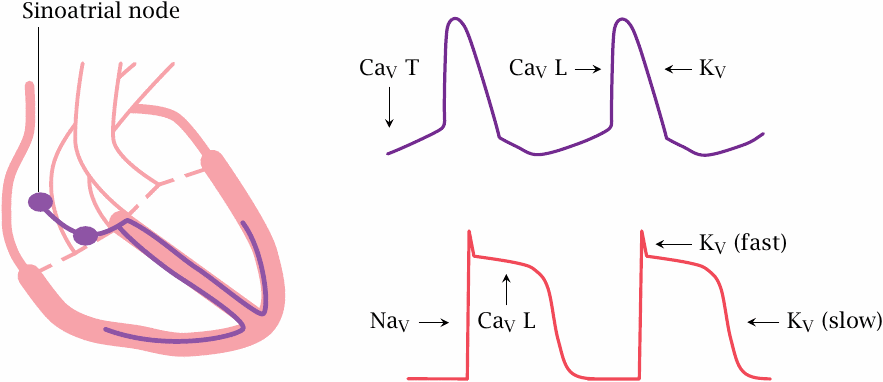
The interplay of sodium and potassium channel controls the action potentials in nerve and skeletal muscle fibers, which are of short duration and often occur with high repetition rates. In contrast, action potentials in the heart last much longer and occur with lower frequency, and their control involves voltage-gated channels for calcium in addition to those for sodium and potassium.
The heart contains two types of excitable cells: those in the excitation-conduction system generate action potentials spontaneously and periodically, whereas the worker muscle cells stay put until they are ordered otherwise, much like the muscle fibers in skeletal muscle. The latter is true at least of a healthy heart; however, in certain forms of cardiac arrhythmia, worker cells create havoc by firing autonomously, and drug treatment will then aim at silencing them again.
The various parts of the excitation-conduction system are hierarchically organized. In a healthy heart, action potentials are generated in the sino-atrial node, which is the topmost center of the excitation-conduction system. From there, the action potentials travel to the atrioventricular node and then spread across the remainder of the system and to the worker cells.39
In the sinoatrial node, T type CaV channels open slowly and spontaneously at the resting potential, causing a gradual depolarization. Once this depolarization reaches the firing level of the faster and more abundant L type CaV channels, these open rapidly and trigger an action potential.
In the worker muscle cells, the action potential is initiated by NaV channels and sustained for the duration of the contraction through CaV channels. Repolarization is mediated by L channel inactivation and by opening of some fairly tardily responding KV channels. Among these, the so-called hERG channels have a prominent role as drug targets and antitargets (see section 6.6).
| 6.4 |
Measuring ion fluxes across single channels |
One of the allures that ion channels hold for many researchers is the ease and detail with which the behavior of individual channel molecules can be studied. Here, we take a brief look at how this is done.
| 6.4.1 |
Planar lipid bilayers |
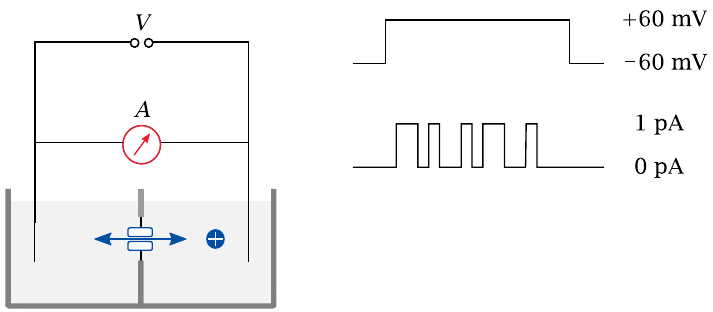
In this experimental setup, a single lipid bilayer is “painted” across a small aperture between two buffered reservoirs. Purified channel molecules, typically solubilized with some non-denaturing detergent, are introduced into one reservoir. Membrane proteins prepared in this way tend to spontaneously insert again into lipid bilayers when offered the opportunity; the right amount of protein sample that will cause just one channel molecule to find and insert into the membrane patch is determined by trial and error.
Electrodes are inserted into the buffer reservoirs on both sides of the bilayer. Voltage is applied, and the opening and closing of the channel can be observed, typically in the form of discrete jumps of constant magnitude that is proportional to the conductivity of the channel molecule.
The buffer solutions usually contain salts in high concentration (~1 M), since this will yield greater currents in response to channel opening. Even then, the currents are typically on the order of only 1 pA.40 Additional compounds—drugs, for example—can be introduced into either chamber to examine their effect on the channel from either end.
| 6.4.2 |
The patch clamp technique |
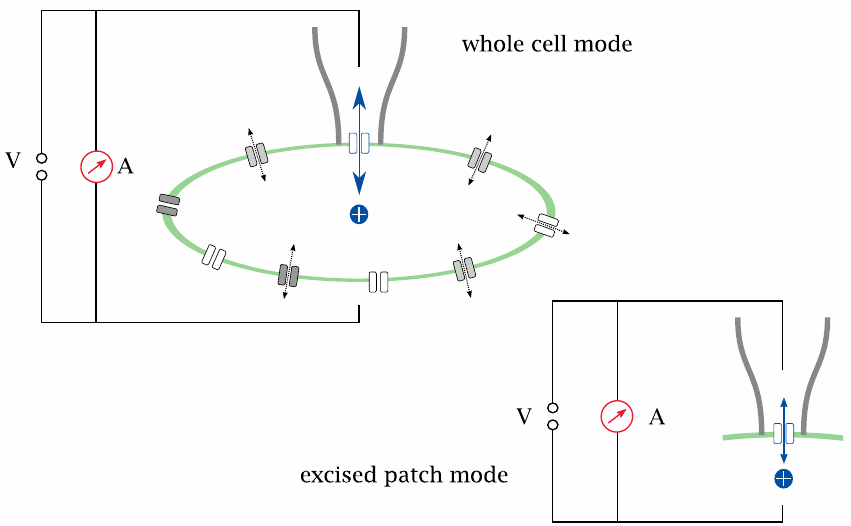
In the patch clamp technique, the channel is observed in its natural environment within the cell membrane; therefore, interactions with regulatory proteins or other effectors are preserved. Isolated observation of a single channel, or of a small number of channels, is accomplished by gently lowering a glass micro-pipette onto the cell surface, such that it forms a seal with the cell membrane. All current through the pipette must then flow through the membrane patch delineated by the pipette aperture, and through any channels this patch happens to contain.
Patch clamp experiments can be performed either in whole-cell mode or in excised-patch mode. In whole-cell mode, the current has to pass the membrane twice in order to close the circuit: once across the sealed patch, and once across the remainder of the cell surface. However, the latter contains very many channel molecules working in parallel, causing negligible ohmic resistance; therefore, the current is controlled almost exclusively by the channels in the patch. In excised-patch mode, the pipette is withdrawn after seal formation; the cell membrane ruptures, with the pipette holding on to the sealed patch of membrane. This makes it possible to experimentally vary the buffer milieu on both the extracellular side and the intracellular side of the membrane.
Channels of different specificities will usually be present in the membrane patch, and in order to selectively observe one species, some others may need to be suppressed, either through addition of specific inhibitors, or through elimination of their specific substrate ions from the buffer. For example, sodium and potassium channels may be suppressed by replacing Na+ and K+ with tetraethylammonium ions; this trick can be used to selectively detect calcium currents.
| 6.5 |
Drug effects on voltage-gated sodium channels |
Sodium channels play a crucial role in the propagation of action potentials, and thus they are a good target when inhibition of nerve conduction is desired, as it is in local anesthesia. In addition, inhibitors of sodium channels are also used in several other situations that require neural excitability to be dampened.
| 6.5.1 |
Fast and slow channel block |
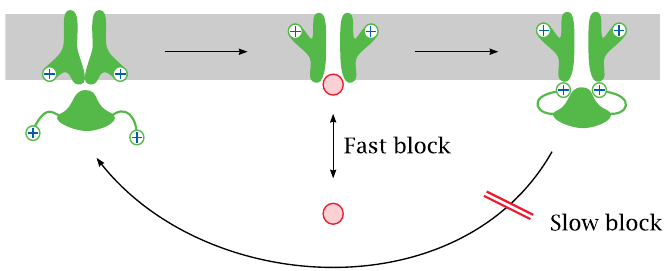
Channel-blocking drugs may cause either fast or slow blocks. A fast block occurs when a drug reversibly binds within the channel lumen and obstructs it.
A slow block is observed when a drug binds to the inactivated state of the channel and delays its reactivation. Both fast and slow blocks may be use-dependent, which means that the channel needs to be activated in order to allow the drugs to access their binding sites and apply the block.
| 6.5.2 |
Diethylamine and phenol resemble parts of the lidocaine molecule |
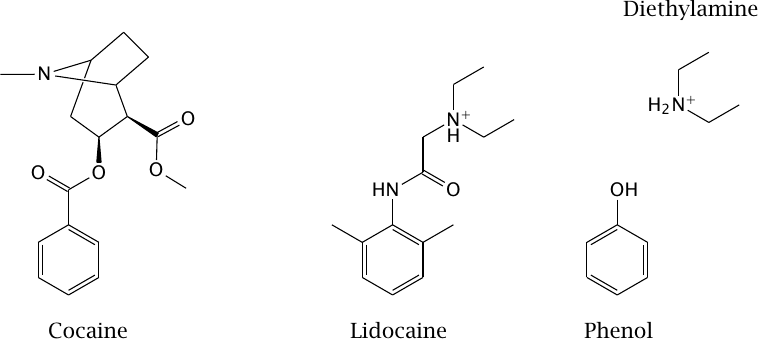
The drug lidocaine is used for local anesthesia and in the treatment of certain types of cardiac arrhythmias. It causes both slow and fast block at the NaV channel. The model compounds diethylamine and phenol resemble parts of the lidocaine molecule; as we will see in the next slide, they also mimic partial activities of it.
Cocaine also blocks sodium channels and can be used for local anesthesia,41 but it is no longer used in this application. This activity of cocaine is unrelated to its psychotropic effect that is caused in dopaminergic synapses (slide 6.14.3).
| 6.5.3 |
Effects of diethylamine and of phenol on NaV channel conductance |
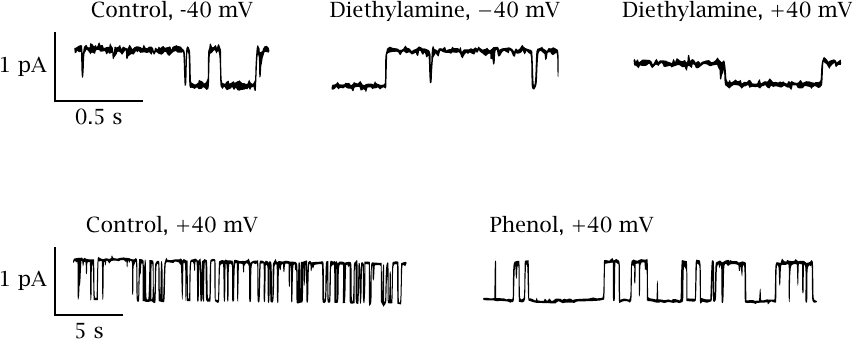
This slide shows a series of single channel recordings on NaV channels in planar lipid bilayers. The channels were biased towards the open state with batrachotoxin (see next slide). Diethylamine reversibly blocks the open state; the block is more pronounced if the membrane potential pushes the drug into the channel (as is the case at +40 mV). Individual binding and dissociation events occur faster than can be resolved by the recording electronics, which therefore averages them out and reports an apparent decrease in the conductance. Such behavior constitutes a fast block.
In contrast to diethylamine, phenol measurably extends the time intervals of channel inactivity but does not alter the observed conductivity of the open state. This effect represents a slow block. Thus, it appears that diethylamine and phenol respectively account for the dual fast and slow block that was reported previously for lidocaine. Figure adapted from [44].
| 6.5.4 |
Drugs and poisons that act on NaV channels |
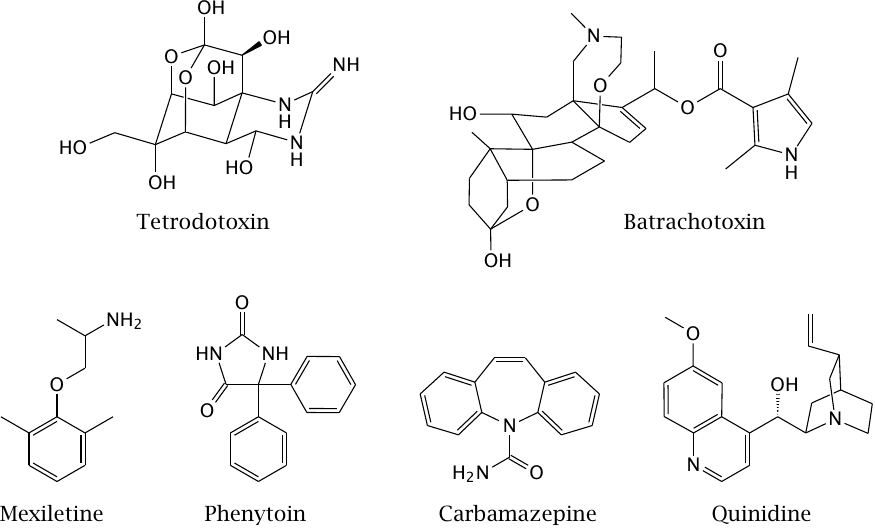
Batrachotoxin, which is found in certain frogs and some other animal species, interacts with the channel from within the lipid bilayer and activates it. It has no therapeutic application but is useful in experimental studies on NaV channels, such as the one illustrated in the preceding slide.
All other molecules shown here are NaV channel blockers. Tetrodotoxin is the poison found in pufferfish. Mexiletine is a more metabolically stable analogue of lidocaine; it can be applied orally and is used in some forms of cardiac arrhythmia, as is quinidine. Carbamazepine is used to treat epilepsy and some other neurological and psychiatric diseases.
Phenytoin is also used in epilepsy. In addition to blocking sodium channels, it also affects some types of potassium and calcium channels. These latter effects may give rise to cardiac arrhythmias but also contribute to its antiepileptic efficacy.
| 6.6 |
Pharmacology of potassium channels |
Among the various subtypes of voltage-gated potassium channels, the most important drug target is the so-called hERG channel, which mediates the repolarization and terminates the action potential (see slide 6.3.5). Inhibition of hERG channels with drugs such as sotalol (slide 6.6.3) and amiodarone (slide 7.3.6) extends the duration of the action potential and of the subsequent refractory period. This is useful in some forms of cardiac arrhythmia. However, hERG channel inhibitors also may trigger their own peculiar forms of arrhythmias, sometimes leading to sudden cardiac death. Blockade of hERG channels is also a remarkably frequent occurrence with drugs that primarily act on different targets entirely; an example is terfenadine [45], which accordingly has been supplanted by its metabolite fexofenadine (see slide 4.2.2). Novel drug candidates are nowadays routinely screened for this particular side effect.
In addition to voltage-gated potassium channels, there are several other families of potassium channels. K+ leak channels, which are always open, dominate the resting potential, whereas other K+ channel types are regulated by various ligands, including Ca++ or G protein βγ-dimers (see slide 5.3.1).
A pharmacologically important K+ channel, which belongs to the Kir family,42 is associated with and controlled by another membrane protein, the sulfonylurea receptor. This receptor functions as an ATP sensor; when bound to ATP, it closes the channel. The complex comprising the channel and the receptor is referred to as the KATP channel.
| 6.6.1 |
KATP channels regulate the tone of smooth muscle cells |
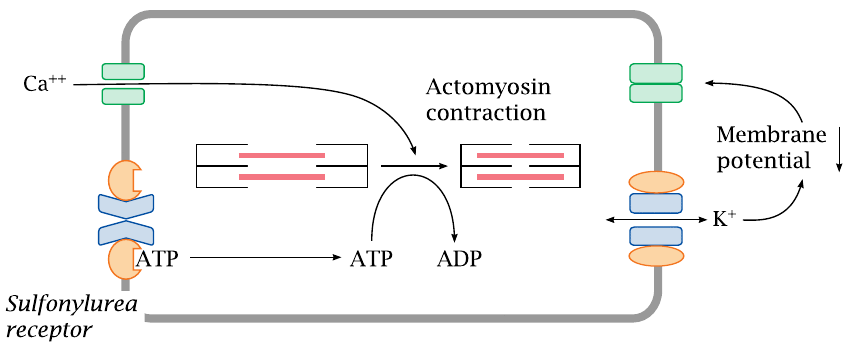
In vascular smooth muscle cells, the KATP channel serves to regulate the strength of contraction. Sustained contraction of the cell will deplete ATP. This will promote dissociation of ATP from the sulfonylurea receptor and open the connected Kir channel. The increase in K+ permeability will hyperpolarize the membrane, that is, lower the membrane potential, and thus inhibit the activation of CaV channels; the overall effect will be inhibition of contraction. If a smooth muscle cell is exhausted and depleted of ATP, opening of the KATP channels lets it “tune out” calcium signals and suspend contraction until it has caught its breath and replenished ATP.
Drugs such as diazoxide (slide 6.6.3) that counteract the effect of ATP on the sulfonylurea receptor will keep the Kir open and promote relaxation of vascular smooth muscle cells. This is an effective means to lower blood pressure.
| 6.6.2 |
KATP channels in pancreatic β cells regulate insulin secretion |
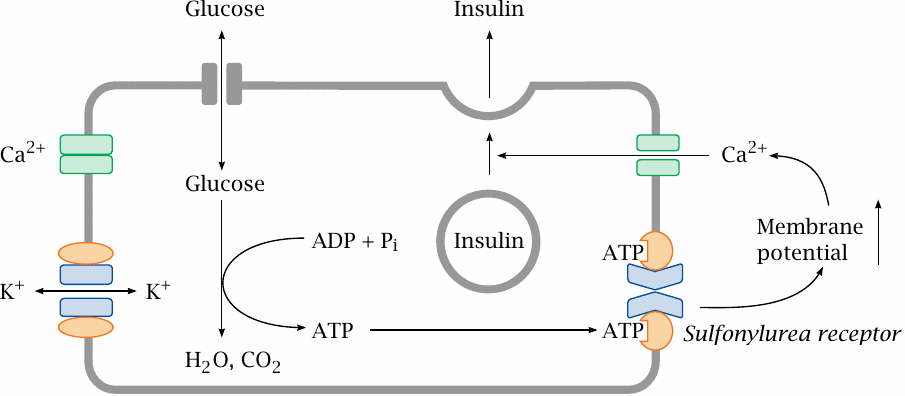
KATP channels also occur in the β cells of pancreatic islets, where they regulate the release of insulin in response to changing blood glucose levels. While most cells require insulin for the uptake of glucose, the islet cells themselves do not; therefore, the intracellular glucose levels will rise and fall with blood glucose.
When blood glucose is high, the islet cells will take up more of it and then degrade it. This produces ATP, which binds the sulfonylurea effector, closes Kir, increases the membrane potential and thereby promotes calcium-mediated insulin release. Pharmacological activation of the sulfonylurea receptor in order to increase insulin secretion is a useful strategy in type 2 diabetics, who, in contrast to type 1 diabetics, retain the ability to produce their own insulin.
Note that the intended effects on the KATP channel differ between the two applications: in type 2 diabetics, we want the channel to close, whereas in hypertension we want it to open. This leads to mutual side effects of antidiabetic and antihypertensive therapy. However, the channel molecules found on β cells and smooth muscle cells are slightly different, which may be exploited by selective drugs to reduce this mutual interference.
| 6.6.3 |
Drugs that act on K+ channels |
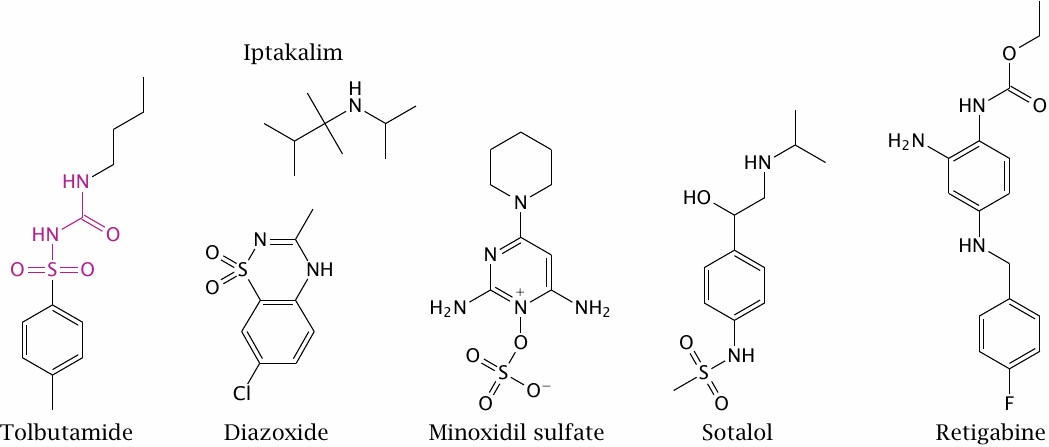
Tolbutamide is a sulfonylurea receptor agonist and closes the associated Kir channel. It promotes insulin secretion from pancreatic β cells and is used for oral therapy in type II diabetics. (In the structure of tolbutamide, the sulfonylurea moiety is highlighted.)
Diazoxide and minoxidil sulfate are sulfonylurea receptor antagonists. They open Kir channels on vascular smooth muscle cells, which induces relaxation and lowers the blood pressure. The experimental drug iptakalim reportedly opens Kir channels in the vasculature but closes those in pancreatic β cells.
Sotalol inhibits KV channels of the hERG type; this inhibition delays repolarization and prolongs the action potential. The drug also blocks β-adrenergic receptors. In combination, these two effects can be useful in certain forms of cardiac arrhythmias. Retigabine activates neuronal KCNQ channels, which also belong to the KV family. It reduces neuronal excitability and is used to treat epilepsy.
| 6.7 |
Pharmacology of calcium channels |
Calcium channels have a prominent role in controlling the rhythm and contraction of the heart (see slide 6.3.5). Drugs that affect T type CaV channels will preferentially act on the excitation-conduction system, whereas sodium channel blockers such as lidocaine will act mostly on the worker cells. Drugs that act on K+ channels or L type calcium channels will affect both cell types.
Calcium channels also control the activity of vascular smooth muscle and the release of neurotransmitters from presynaptic terminals (see slide 6.1.3). All of these physiological effects have found pharmacological applications.
| 6.7.1 |
Two calcium channels control the contraction of striated muscle cells |
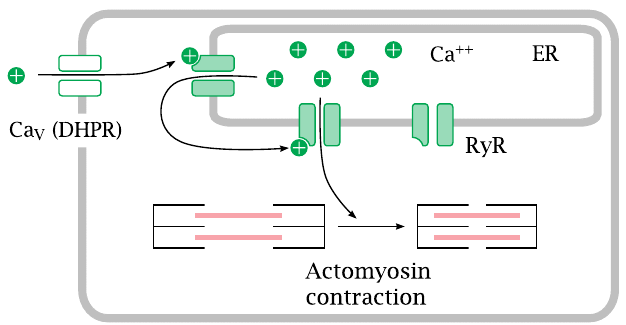
Voltage-gated calcium channels of type L are also referred to as dihydropyridine receptors (DHPRs). They control contraction both in the heart and in skeletal muscle cells.
When an action potential arrives and opens the channel, calcium flows in and opens the ryanodine receptor (RyR), a calcium-activated calcium channel that sits in the membrane of the endoplasmic reticulum. The large secondary wave of calcium released by the open RyR then triggers actomyosin contraction.
| 6.7.2 |
Entry of Ca++ through the DHPR is necessary in the heart, but not in skeletal muscle cells |
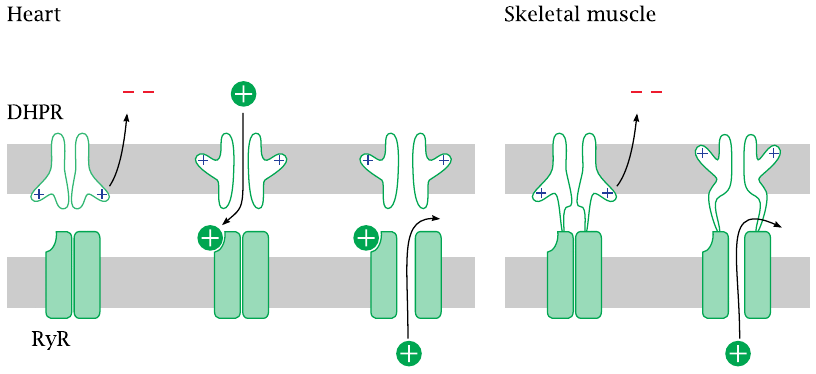
There exists a surprising difference in the interaction of the two calcium channels between heart and skeletal muscle. In the heart, activation of the ryanodine receptor (RyR) requires actual influx of Ca++ through the dihydropyridine receptor (DHPR), which opens during an action potential.
In skeletal muscle, the two channels are hooked up directly to each other. Through this connection, the conformational change that occurs in the DHPR upon membrane depolarization is communicated to the RyR and causes it to open. Therefore, no actual influx of extracellular Ca++ is necessary—contraction can still occur even if the DHPR channel is plugged up, as long as it still performs its conformational pantomime of gate opening. Therefore, DHPR channel blockers will not interfere with contraction of skeletal muscle.
DHPR channels are also found in vascular smooth muscle. As in heart muscle cells, they don’t couple directly to ryanodine receptors. Therefore, DHPR blockers will affect the function of the heart and the blood vessels but not of the skeletal muscles.
| 6.7.3 |
Inhibitors of voltage-gated calcium channels |
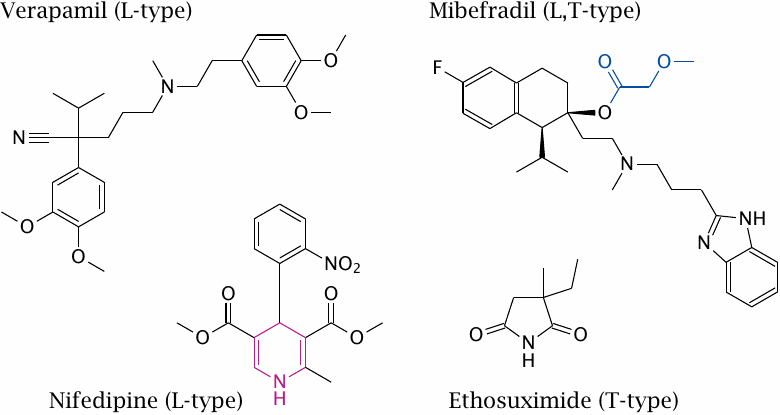
Nifedipine and verapamil both inhibit L type CaV channels. As stated above, these channels are also called dihydropyridine receptors (DHPRs), after the dihydropyridine moiety that occurs in some inhibitors. In the structure of nifedipine, the dihydropyridine group is highlighted.
Mibefradil inhibits both L type and T type calcium channels. Its effect on L type channels has been ascribed to an active metabolite in which the highlighted substituent is hydrolytically removed. Replacement of this group with a cyclopropyl moiety reportedly produces a selective T type channel blocker [46].43
Ethosuximide selectively blocks specific T type CaV channel variants that are found on nerve cells in the brain. The drug is used in specific forms of epilepsy.
| 6.7.4 |
Structure of ω-conotoxin, an inhibitor of N-type CaV channels |
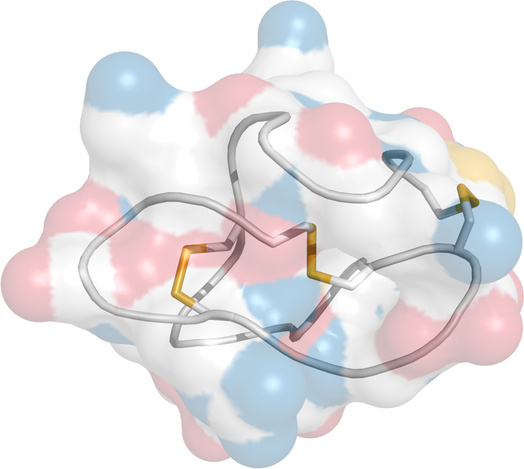
N type channels are those CaV channels that are found on presynaptic nerve terminals and mediate neurotransmitter release (see slide 6.1.3).
The ω-conotoxins, produced by cone snails, selectively block N type channels. Some conotoxins are of interest in the experimental study or treatment of pain [48]. The figure shows ω-conotoxin CVID. Oxygen is rendered in red, nitrogen in blue. Disulfide bonds are rendered in yellow.
| 6.7.5 |
Agonists of transient receptor potential (TRP) channels |
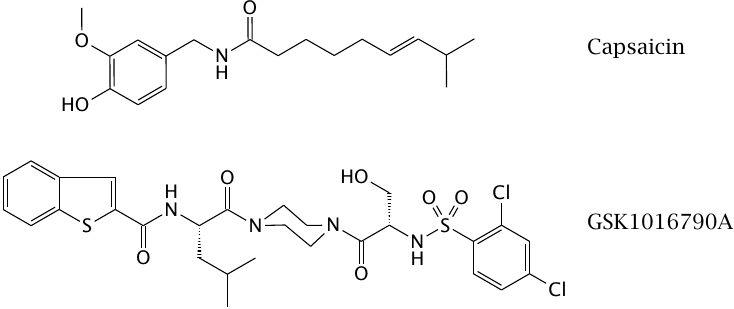
Transient receptor potential channels conduct calcium as well as other cations.44 They respond to various types of physical stimuli, such as heat or mechanical tension, and are found in various types of sensory cells, such as those for sound waves in the inner ear and for heat in the skin and mucous membranes. They may also be activated chemically; for example, the TRPV1 receptor is activated both by heat and by capsaicin, the active ingredient of chili peppers—which validates the ambiguity of our notion of “hot” food.
The experimental drug GSK1016790A activates the mechanosensitive TRPV4 channel, which is found in the circulation and acts as a blood pressure sensor. Activation of this receptor will feign an elevation of blood pressure to the autonomic nervous system and trick it into issuing a reflex to relax the blood vessels and lower the pressure. However, activation of the same receptor also causes leakiness of capillary endothelia [49], which makes drugs of this type unsuitable for therapy.
| 6.8 |
Na+/K+-ATPase maintains the ion gradients at the plasma membrane |
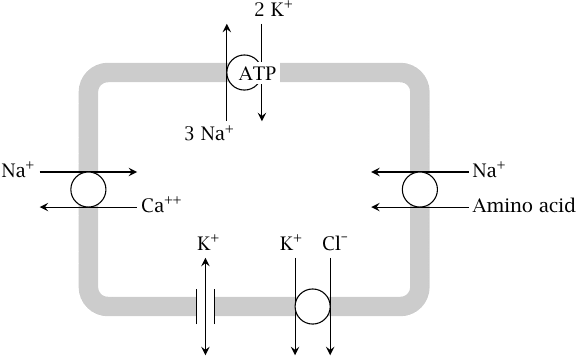
We had seen earlier that, with the exception of calcium, the ion concentrations are not changed significantly by the fluxes that occur during single action potentials. However, over time, the repetitive firing of action potentials will wear down the ion gradients. Therefore, all ion gradients must be maintained through active transport that ultimately depends on ATP expenditure.
The key molecule in maintaining the cellular ion concentration gradients is Na+/K+-ATPase, which keeps up the gradients of the two major cations; secondary active transport coupled to Na+ or K+ sustains the other ion gradients. The high extracellular sodium concentration drives calcium antiport as well as symport of amino acids and other metabolites. Potassium symport drives the export of chloride from the cell. Potassium leak channels maintain the negative-inside resting potential potential.
| 6.8.1 |
Functional cycle of Na+/K+-ATPase |
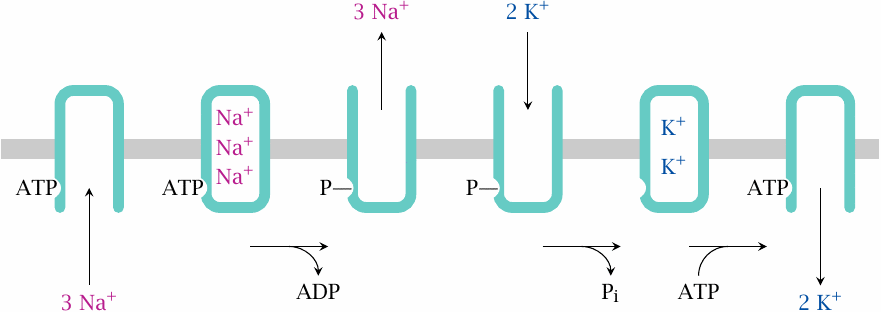
The functional cycle of Na+/K+-ATPase involves the following stages:
- The ATP-bound form of the enzyme is open to the cytosol and accepts three Na+ ions. Bound Na+ becomes occluded inside the enzyme.
- The occluded enzyme phosphorylates itself and releases ADP. The phosphorylated molecule everts and releases the bound Na+ to the extracellular space.
- Two K+ ions bind from the outside and become occluded. Concomitantly, phosphate is hydrolyzed.
- ATP binds, which opens the enzyme to the intracellular side. This releases K+ and restores the enzyme to its initial state.
This sequence of events has been pieced together from numerous experimental studies. The following slides present some of the key experiments.
| 6.8.2 |
Na+/K+-ATPase activity as a function of KCl and NaCl concentrations |
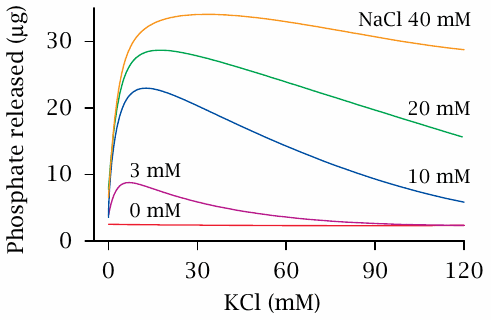
Neither NaCl nor KCl alone elicit significant ATP cleavage activity. Maximal ATP hydrolysis is observed at approximately equal concentrations of KCl and NaCl. In this experiment, the enzyme had been extracted from membranes; therefore, only ATP hydrolysis, but no transport phenomena were observed. This early result gave the enzyme its name. Figure prepared from original data in [50].
| 6.8.3 |
Effects of Rb+ and of Na+ on the phosphorylation state of Na+/K+-ATPase |
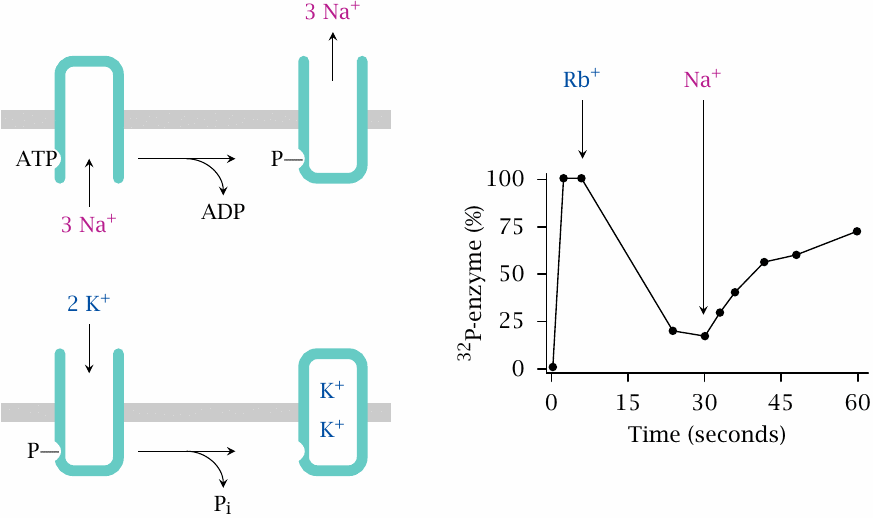
In this experiment, the enzyme was incubated with 32P-labeled ATP. At time zero, only Na+ is present, and phosphorylated enzyme accumulates; therefore, sodium induces autophosphorylation of the enzyme, but not its dephosphorylation.
Addition of Rb+, which mimics K+, dephosphorylates the enzyme; Na+ added in excess over Rb+ phosphorylates it again. Therefore, binding—and, presumably, transport—of sodium is associated with phosphorylation, whereas binding of K+ is associated with dephosphorylation. This explains why both are required for continuous, catalytic cleavage of ATP. Figure prepared from original data in [51].
| 6.8.4 |
ATP is required to release Rb+ from tight binding to Na+/K+-ATPase |
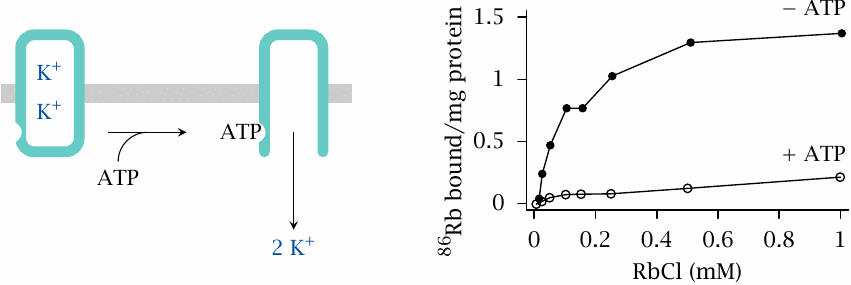
As stated before, Rb+ is a substitute for K+.45 Tight binding of Rb+ is interpreted as occlusion of the ion within the enzyme. Figure prepared from original data in [52].
| 6.8.5 |
Structures of the Na+/K+-ATPase inhibitors ouabain and digitoxin |
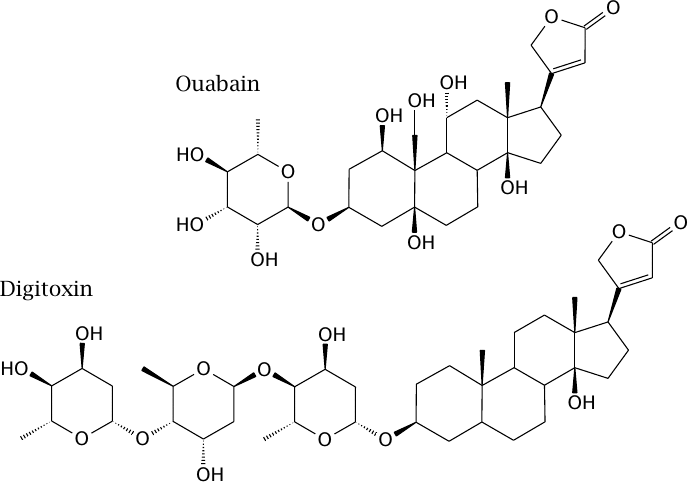
As discussed earlier (see slide 6.8), the Ca++ gradient across the cytoplasmic membrane is sustained by the Na+ gradient, which in turn is maintained by Na+/K+-ATPase. Therefore, inhibition of this enzyme will increase the intracellular calcium level. In muscle cells, this will tend to increase the force of contraction.
The Na+/K+-ATPase inhibitors digitoxin and the related drug digoxin are isolated from the foxglove plant (Digitalis purpurea). They are used clinically in the treatment of cardiac insufficiency to augment the contractile force of the heart muscle. Ouabain has the same effect but is inconvenient for clinical use because of rapid elimination; it is mostly used experimentally.
| 6.9 |
Overview of synaptic transmission |
In a chemical synapse, the electrical signals traveling around the nervous system are transiently converted to chemical signals. The transmitter substances used in different types of nerve cells are quite diverse, and specific cell types or neural subsystems can be addressed in a more selective manner than is usually possible with the drug targets we have considered so far. Accordingly, most drugs that act on the central nervous system target chemical synapses.
| 6.9.1 |
Function of a chemical synapse |
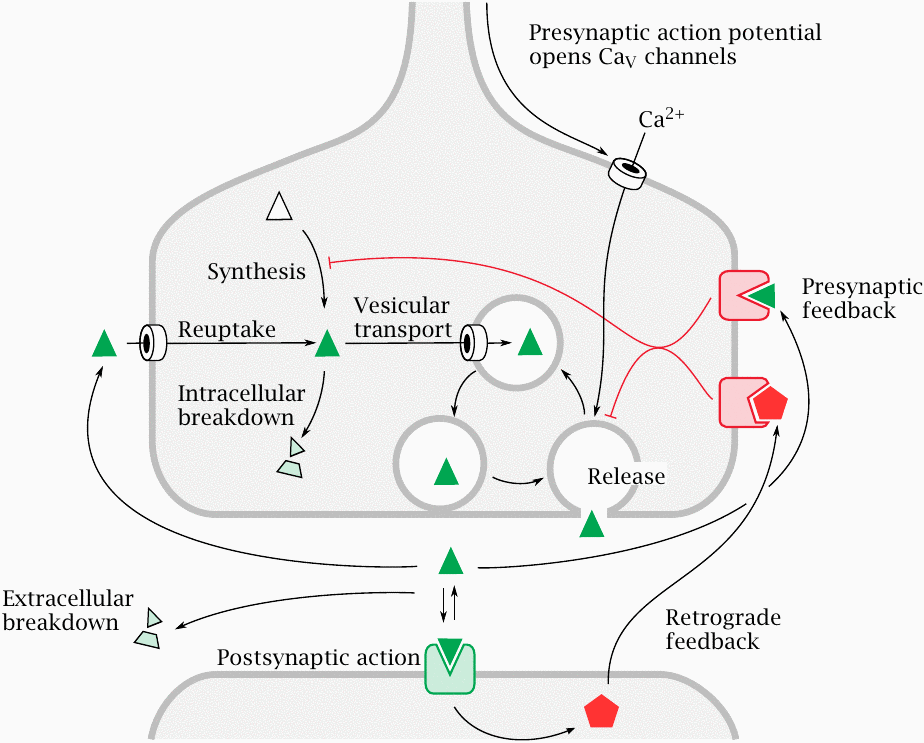
Synaptic transmission is initiated by N-type CaV channels, which are activated by action potentials arriving via the axon from upstream. Ca++ ions enter the nerve terminal and trigger exocytosis of vesicles loaded with neurotransmitter. Released transmitter molecules bind and activate postsynaptic receptors. Depending on the functional properties of these receptors, the postsynaptic cell is either excited or inhibited.
In response to stimulation by transmitters, a postsynaptic cell may release mediators that exert retrograde negative feedback on the presynaptic cell.46 The transmitter itself can also cause negative presynaptic feedback, through receptors that usually differ from those found on the postsynaptic membrane.
A short while after its release, the transmitter is inactivated; this may occur through either extracellular breakdown or presynaptic reuptake. Transmitter vesicles are recycled and loaded with transmitter through active transport. The pool of transmitter for loading into the vesicles is controlled by the rates of transmitter synthesis, degradation, and reuptake.
All these processes provide drug targets and create opportunities for pharmacotherapy, and we will see one or more example drugs for each of them.
| 6.9.2 |
Summation of postsynaptic potentials |
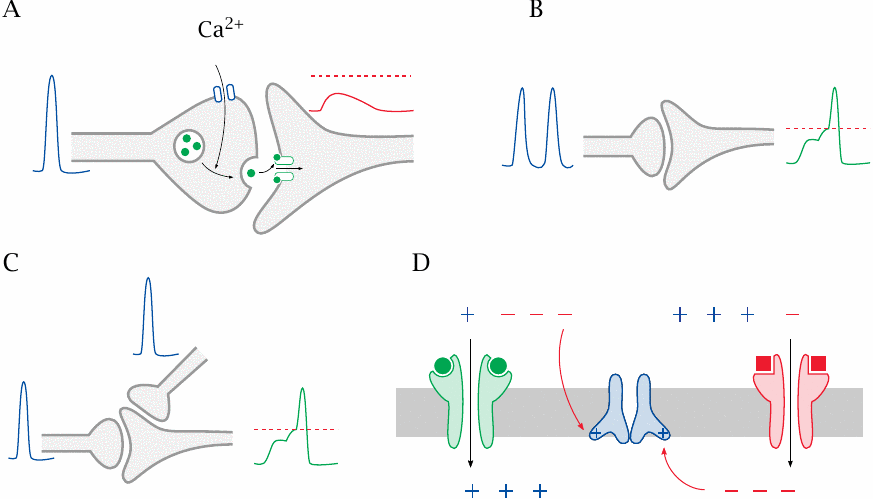
We have seen before that a given nerve cell may receive input from a large number of other nerve cells upstream. If each nerve cell were to trigger an all-out action potential in response to every single stimulus from upstream, all cells would very soon be mutually stimulating each other to exhaustion. Therefore, a nerve cell does not respond all out to a single upstream stimulus but instead requires multiple input stimuli before firing.47 This slide explains how that works.
- (A)In excitatory synapses between nerve cells, a single presynaptic action potential causes a localized partial depolarization, the excitatory postsynaptic potential (EPSP). A single EPSP will remain below the firing level and not trigger a postsynaptic action potential.
- (B)An action potential may form if a single synapse fires repeatedly before the EPSP dissipates; this is known as temporal summation.
- (C)Alternatively, in spatial summation, the EPSPs caused by neighboring synapses firing at the same time may combine to trigger an action potential.
- (D)Inhibitory synapses, which often employ chloride channels, cause inhibitory postsynaptic potentials (IPSPs), which counteract the activation of voltage-gated channels by EPSPs in the vicinity.
The firing level that may or may not be reached by EPSPs is the threshold voltage at which the neighboring voltage-gated ion channels will be activated and set off a self-sustaining wave of depolarization, that is, an action potential (see slide 6.3.3). Also note that, unlike in this cartoon, summation of more than just two EPSPs will be required to actually trigger a postsynaptic potential.
| 6.9.3 |
Neurotransmitter receptor families |
-
Ligand-gated channels
- Cys-loop family
- Glutamate receptors
- Purine P2X receptors
- G protein-coupled receptors
The most straightforward drug targets, and also the most widely used ones, are the neurotransmitter receptors on the postsynaptic membranes. The feedback receptors on the presynaptic membranes are sometimes targeted as well. All of these receptors belong to two major functional classes:
- 1.Ligand-gated channels, also referred to as ionotropic receptors, open in response to the binding of their cognate transmitters and thereby directly change the potential at the postsynaptic membrane. This mode of action is very rapid. Ligand-gated channels fall into three distinct structural families, the members of which—in the above order, from (a) to (c)—have five, four, and three subunits, respectively.
- 2.G protein-coupled receptors affect the membrane potential in various and somewhat more roundabout ways. One important signaling mechanism is through adenylate cyclase, cAMP, and cyclic nucleotide-gated (CNG) channels. The CNG channels conduct all major cations; on balance, they will depolarize the membrane and promote formation of postsynaptic action potentials.
Most transmitters have multiple receptor types, and some, like glutamate and acetylcholine, have receptors among both the ligand-gated channels and the GPCRs.
| 6.10 |
Structure, function, and pharmacology of cys-loop receptors |
Among the ligand-gated channels, this family contains the most clinically important drug targets, and it also contains the most widely studied ionotropic receptor molecule, namely, the nicotinic acetylcholine receptor.
| 6.10.1 |
Structure of the nicotinic acetylcholine receptor |
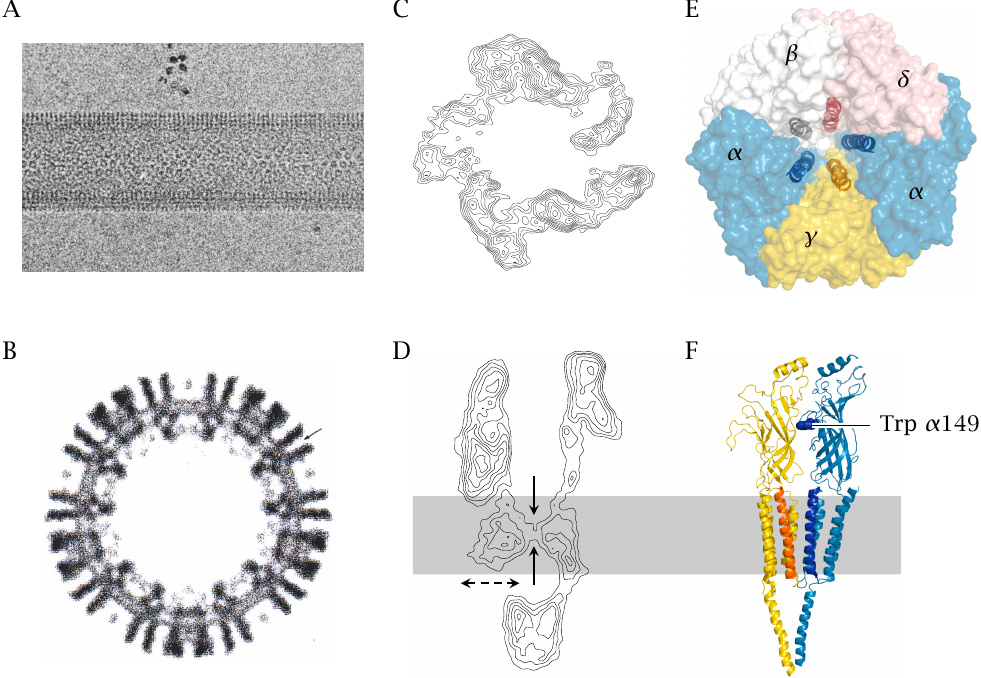
The nicotinic acetylcholine receptor (NAR) conducts cations, in a fairly nonselective way; Na+, K+ and Ca++ can all get across. It depolarizes the postsynaptic membrane.48
The structure shown here has been worked out with electron crystallography, which is sort of a hybrid between conventional crystallography and electron microscopy. The individual panels show the following:
- A, B:
- Electron micrographs of tubular synaptic membrane particles isolated from Torpedo electric organs. The cross-section in B already shows the major structural features of the receptor. The formation of dense, regularly packed arrays as seen in A allows the collection of in-phase electron diffraction patterns, from which higher resolution contour maps can be constructed.
- C, D:
- Contour maps obtained by electron crystallography. In the side view (D), a constriction within the bilayer is clearly visible. The receptor has an intracellular domain with lateral openings.
- E:
- High resolution structural model, top view. The receptor consists of five subunits, each of which contributes one helix to the gate located at the level of the lipid bilayer.
- F:
- Side view of the structural model. Only one of the α chains and the adjacent γ chain are shown. The tryptophan residue at position 149 of the α chain is part of one of the two acetylcholine binding sites, which are located at the α-γ and the α-δ interfaces, respectively.
The subunit composition illustrated here applies to nicotinic receptors in neuromuscular synapses. Receptors at other anatomical sites differ in subunit composition, and some drugs discriminate between these different receptor types. Panels A–D courtesy of Nigel Unwin; E and F rendered from 2bg9.pdb.
| 6.10.2 |
Trapping the nicotinic acetylcholine receptor in the open state |
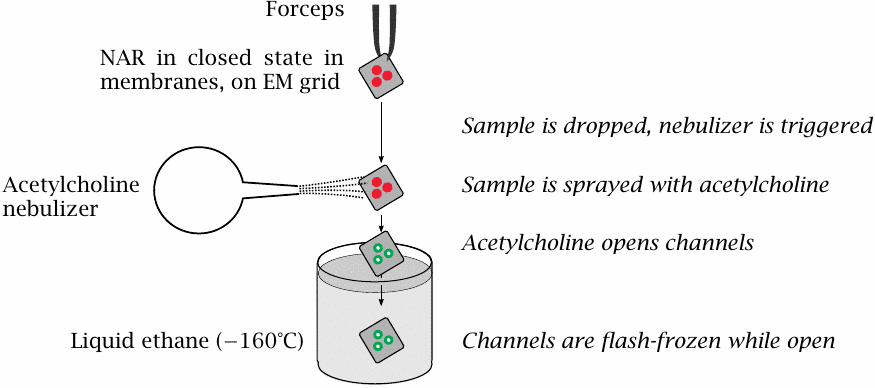
Nigel Unwin managed to study the structure of the nicotinic acetylcholine receptor both in the open and the closed state. Now, considering that the open state has a lifetime of only a few milliseconds, how did he manage to prepare it, and then stabilize it so that it could be imaged? This slide explains his experimental setup.
- The membrane vesicles containing the receptors, in 2D crystal arrangement and in the closed state, are mounted on an EM sample support and held in a forceps.
- When the sample is dropped, it passes through a stream of vaporized acetylcholine. Binding of acetylcholine opens the channels. Immediately thereafter, the sample plunges straight into cold, liquid ethane, and the channels are shock-frozen in the open state.
- Data acquisition for electron crystallography is performed at low temperatures, so as to preserve the receptor in its open state.
Rather brilliant, isn’t it? One of my all time favorite experiments.
| 6.10.3 |
Photoaffinity labeling of the acetylcholine binding site |
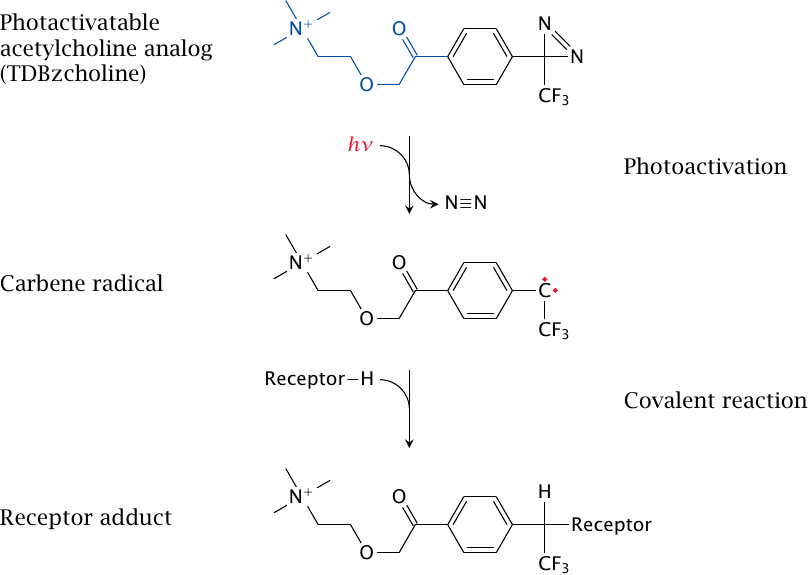
The binding sites of acetylcholine in the receptor have been identified not by 3D-structural methods but by affinity labeling and protein chemistry. Multiple studies using different affinity probes have revealed different features of the binding sites; one such study is summarized here as an example.
The probe used in this experiment, 3H-TDBzcholine, contains a moiety that resembles acetylcholine (highlighted in blue), which steers it toward the acetylcholine binding site of the receptor. It is radioactively labeled and also photoactivatable, which enables it to covalently react with the receptor.
After allowing time for binding of the probe to the receptor, the probe is activated by exposing the sample to UV light. The energy of a photon absorbed by the probe’s aromatic ring induces the release of nitrogen. This leaves behind a highly reactive carbene radical, which immediately reacts with amino acid residues in the vicinity. To identify the labeled amino acid residues, the receptor adduct is then fragmented in a stepwise fashion, as shown in the following slides.
| 6.10.4 |
Isolation of affinity-labeled polypeptide chains and fragments |
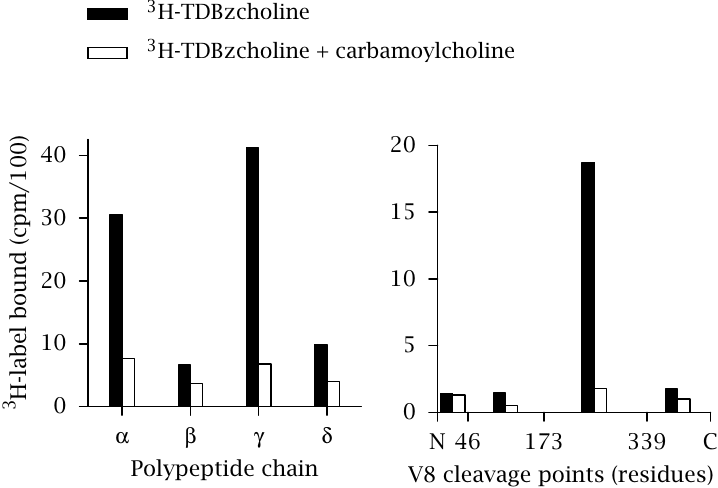
In the first fractionation step, the hetero-oligomeric receptor was simply boiled in SDS and subjected to SDS-PAGE (left panel). The greatest extent of radiolabel incorporation was observed with the α and the γ chain, indicating that these two chains surround the binding site.
In the second step, the labeled chains were fragmented by proteolysis. The protease used here, staphylococcal V8 protease, cleaves specifically after aspartic and glutamic acid residues. The fragments were separated by HPLC and identified by N-terminal sequencing. In the case of the α chain (right panel), the radioactivity was associated with a fragment of 20 kDa that spans residues 174–339.
In both steps, control samples were included that had been exposed to 3H-TDBzcholine in the presence of unlabeled carbamoylcholine in excess. The unlabeled ligand displaced the 3H-TDBzcholine from the acetylcholine binding site and largely suppressed labeling. This confirms that 3H-TDBzcholine indeed lodges within the regular acetylcholine binding site and not in some other, nonspecific binding crevice or pocket.
| 6.10.5 |
Identification of affinity-labeled amino acid residues |
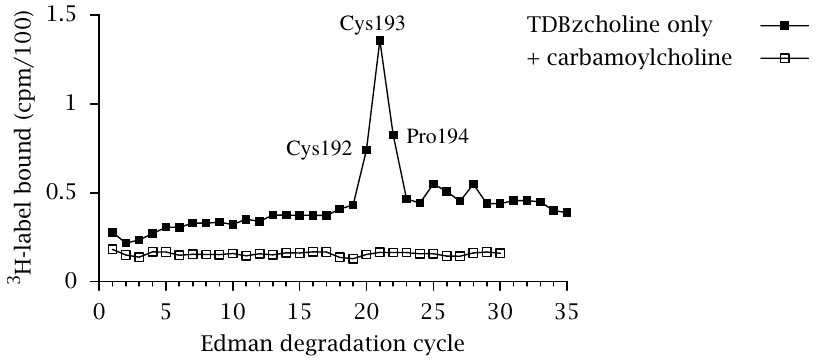
In the final stage of the experiment, the labeled proteolytic fragments were subjected to N-terminal sequencing through Edman degradation in order to identify the individual amino acid residues that had reacted with the label.
In the case of the labeled 20 kDa α-chain fragment shown in the previous slide, residues 192–194 contained the most incorporated label, indicating that they are part of the acetylcholine binding site. Figures in both above slides were prepared from original data in [53].
| 6.10.6 |
Desensitization of the nicotinic acetylcholine receptor |
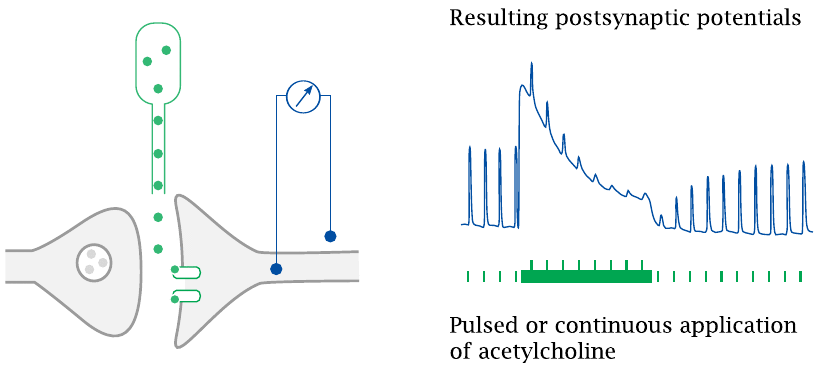
As had been mentioned earlier (slide 2.5.5), the nicotinic acetylcholine receptor undergoes inactivation when bound to its ligand for prolonged periods of time. This property is important in the action mode of the “depolarizing blocker” succinylcholine (slide 6.15.1).
In the experiment illustrated here, acetylcholine was electrophoretically applied to frog muscle neuromuscular synapses, and the resulting postsynaptic potentials were recorded. Repetitive short acetylcholine stimuli evoke postsynaptic potentials of uniform amplitude. When continuous application starts, a strong depolarization occurs, which then declines even in the face of continued transmitter application. The response to superimposed short acetylcholine pulses also declines.
After continuous acetylcholine application is stopped, the response to short acetylcholine pulses recovers over a period of several seconds. Drawn after an original figure in [54].
| 6.10.7 |
Functional states of the nicotinic acetylcholine receptor |
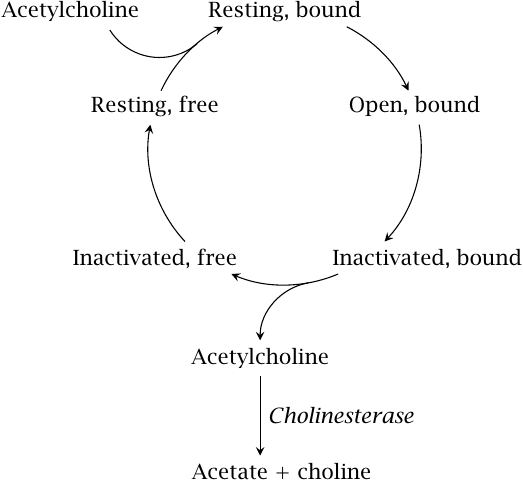
This slide shows a kinetic scheme for acetylcholine binding, receptor activation, and desensitization. When acetylcholine binds, the receptor initially opens, but then quickly inactivates. Opening is faster than inactivation, but the inactive conformation is more stable; therefore, after sufficient time, most receptor molecules will accumulate in the refractory state.
As long as acetylcholine remains bound, the receptor remains inactive. Dissociation of acetylcholine is driven by cholinesterase, which depletes the free transmitter by cleaving it to choline and acetate. Once the bound transmitter molecule has left, the receptor reverts from the refractory state to the inactive one, from which it can again be activated by binding another acetylcholine molecule.
In contrast to a simple equilibrium reaction, which simply flickers back and forth between its two states, a unidirectional cycle like this one requires an input of energy. With the NAR, this energy is supplied by the hydrolysis of acetylcholine.
| 6.10.8 |
Ionotropic receptors in the cys-loop family |
| Receptor | Ion selectivity | Effect | Comments |
| nicotinic acetylcholine | cations | excitatory | pharmacologically distinct subtypes, various applications |
| 5-HT3 serotonin | cations | excitatory | inhibitors are used to treat emesis |
| GABAA | chloride | inhibitory | major drug target in narcosis, epilepsy, psychoses |
| glycine | chloride | inhibitory | regulates motor activity |
The nicotinic acetylcholine receptor occurs in the CNS, in the autonomic nervous system, and in neuromuscular synapses (motor endplates). We will see some drugs that target this receptor in section 6.15, after having examined the organization of the autonomic nervous system.
Inhibitors of the 5-HT3 serotonin receptor are used to suppress emesis, particularly in cancer patients undergoing chemotherapy. The 5-HT3 inhibitor ondansetron is shown in slide 6.12.2.
The GABAA receptor and the glycine receptor are both chloride channels and therefore, when open, hyperpolarize the postsynaptic cells; they are the most important inhibitory neurotransmitter receptors in the brain. The GABAA receptor is one of the preeminent drug targets in the CNS; agonists of this channel are used in conditions as diverse as epilepsy, psychoses, insomnia, and narcosis. We have already seen several GABAA agonists, such as diazepam and oxazepam (slide 4.2.2) as well as phenobarbital (slide 4.1.4) and thiopental (slide 3.6.9). Some more drugs that target GABAA receptors are shown in slide 6.10.9.
The glycine receptor is less prominent as a drug target; some drugs interacting with it are shown in slide 6.10.10. Nevertheless, it has a crucial role in maintaining a proper balance of excitation and inhibition in the CNS, which is quite strikingly illustrated by the clinical manifestations that result when its function is disrupted. The clinical picture is known as tetanus and consists in sustained maximal, uncontrollable contraction of skeletal muscles, to the point where tendons snap and bones break.49
| 6.10.9 |
Drugs that interact with GABA receptors and transporters |
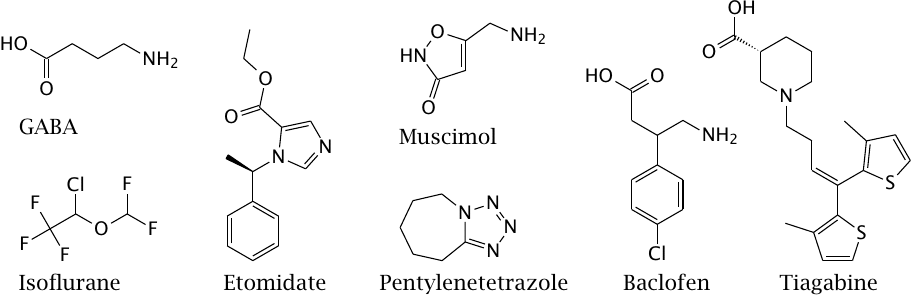
The GABAA and GABAC receptors are ligand-gated channels, whereas the GABAB receptors are GPCRs.
Isoflurane is an inhalation anesthetic, and etomidate is an intravenously applied anesthetic; both are GABAA receptor agonists. Muscimol is an ingredient of fly agaric (a mushroom) and a GABAA and GABAC receptor agonist. Pentylenetetrazol is a GABAA antagonist used to induce seizures in animal experiments.
Baclofen is a GABAB agonist that is used in the treatment of spasticity, for example in patients with multiple sclerosis. Tiagabine is an inhibitor of presynaptic GABA reuptake, which is used in the treatment of epilepsy.
| 6.10.10 |
Drugs that interact with glycine receptors and transporters |
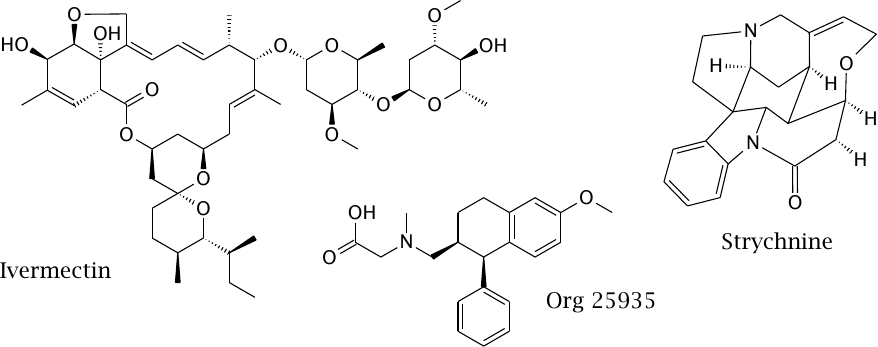
Ivermectin is an allosteric agonist of the glycine receptor. Its main target, however, is a glutamate receptor/chloride channel that occurs in non-vertebrates including some human worm parasites, but not in humans. Accordingly, the drug is used in the treatment of parasite infections.
Strychnine is an alkaloid that acts as an antagonist of the glycine receptor. Once upon a time, it was used as a stimulant, as well as an important ingredient of murder novels. The symptoms of strychnine poisoning resemble those of tetanus. Wikipedia’s page on this subject is well worth a read.
Org 29535 is an inhibitor of glycine reuptake that is being investigated for the treatment of psychosis and addiction.
| 6.11 |
Glutamate receptors |
Glutamate is the major excitatory transmitter in the brain. It has various receptors, both the among ligand-gated channels and GPCRs.
The ionotropic glutamate receptors have four subunits each and thus are structurally distinct from the receptors in the cys-loop family. Ionotropic glutamate receptors are pharmacologically diverse; different types can be selectively activated by the synthetic agonists AMPA, kainate and NMDA, and are accordingly named. Among these receptors, the NMDA receptor is unusual in that one of its four subunits actually binds glycine, not glutamate, and both transmitters are required to activate the receptor. The structures of various glutamate receptor ligands are shown in slide 15.1.1.
While experimental agonists and antagonists with specificity for different glutamate receptors are readily available, compelling clinical applications are scarce [55].
| 6.12 |
Pharmacology of dopamine and of serotonin |
Dopamine belongs to the catecholamines, which also comprise norepinephrine and epinephrine. In their synthesis and degradation, catecholamines share similarities with serotonin. Catecholamines and serotonin also overlap in their pharmacology, and they have prominent roles in regulating mood, vigilance, and voluntary movement. Synapses that use these transmitters are therefore commonly targeted by drug therapy.
| 6.12.1 |
Biosynthesis of the catecholamines and of serotonin |
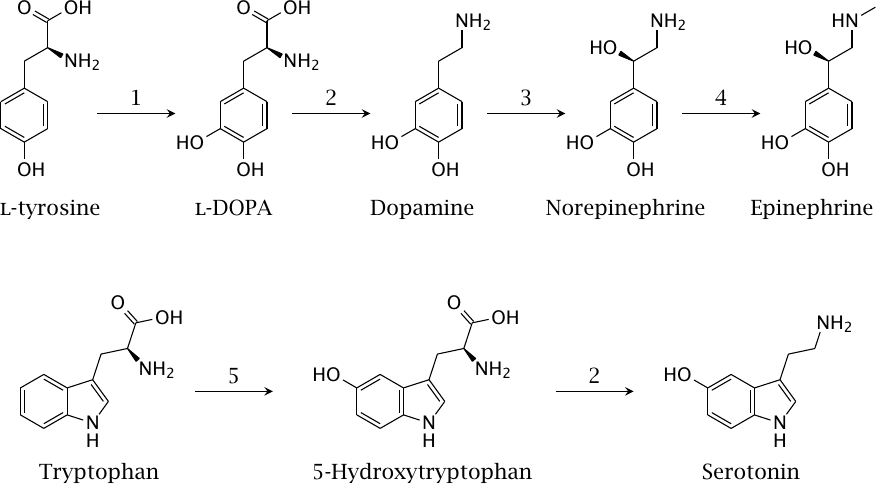
The catecholamines are biosynthetically derived from tyrosine, while serotonin is derived from tryptophan. Hydroxylation and decarboxylation occur in both biosynthetic pathways.
Enzymes: 1, tyrosine hydroxylase; 2, l-aromatic acid decarboxylase or DOPA decarboxylase; 3, dopamine β-hydroxylase; 4, norepinephrine methyltransferase; 5, tryptophan hydroxylase.
| 6.12.2 |
Drugs that interact with dopaminergic and serotoninergic synapses |
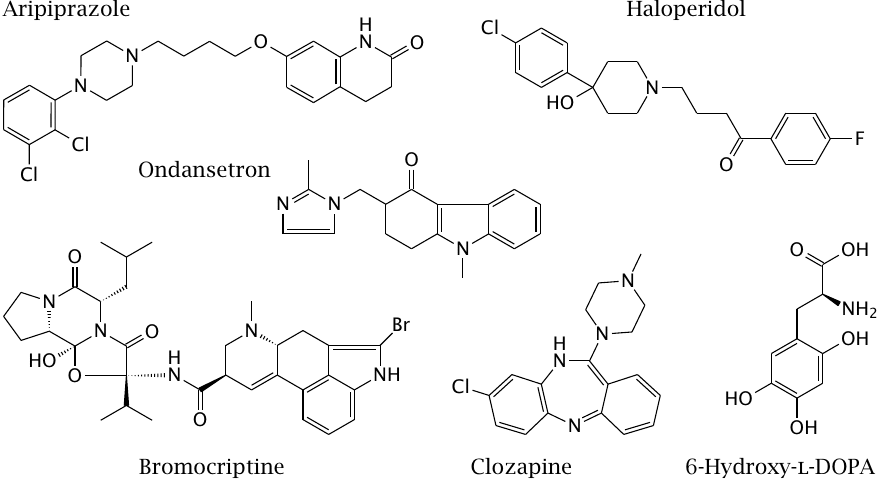
Dopamine and serotonin in the brain are both important in the regulation of mood. Dopamine also participates in the control of body movements, and its deficiency gives rise to Parkinson’s disease (see slide 3.5.5). Accordingly, Parkinson-like symptoms—rigidity, tremor, and slowness of movement—are a common side effect of some dopamine receptor antagonists that are used as antipsychotic drugs.
Haloperidol is a first-generation antipsychotic and a D2 dopamine receptor antagonist with a pronounced tendency to cause Parkinson-like symptoms. Bromocriptine is an agonist at the same receptor. It is used in Parkinson’s disease and to suppress prolactin secretion; this latter application relates to yet another physiological function of dopamine, namely, the regulation of hormone secretion in the hypothalamus and the hypophyseal gland (see section 7.2).
Clozapine is a second-generation antipsychotic with relatively low affinity for D2 receptors but higher antagonistic activity on D3 and D4 dopamine receptors, as well as on 5-HT2A serotonin receptors. Aripiprazole is another second-generation antipsychotic; it is a partial agonist at the D2 receptor and otherwise similar to clozapine. Both of these drugs are much less prone to triggering Parkinson-like symptoms than first-generation drugs like haloperidol.
6-Hydroxy-l-DOPA is not a therapeutic drug but rather a poison. It is transported into dopaminergic neurons by reuptake and damages the cells through inhibition of the respiratory chain. It is sometimes used in research to induce a Parkinson-like condition in experimental animals.
Ondansetron is an antagonist at the 5-HT3 receptor, the sole ligand-gated channel among the serotonin receptors. It is used mostly in the treatment of chemotherapy-induced nausea in cancer patients.
Another major class of drugs that is used to increase postsynaptic stimulation in dopaminergic and serotoninergic synapses are inhibitors of transmitter reuptake. Some examples will be shown later (see slide 6.14.3).
| 6.13 |
The autonomic nervous system |
The autonomic nervous system is the part of our nervous system that is not under conscious control. It controls many physiological functions, and therefore contains many targets for drug therapy. Prominent neurotransmitters in the autonomic nervous system are acetylcholine and the catecholamines epinephrine and norepinephrine.
| 6.13.1 |
Organization of the autonomic nervous system |
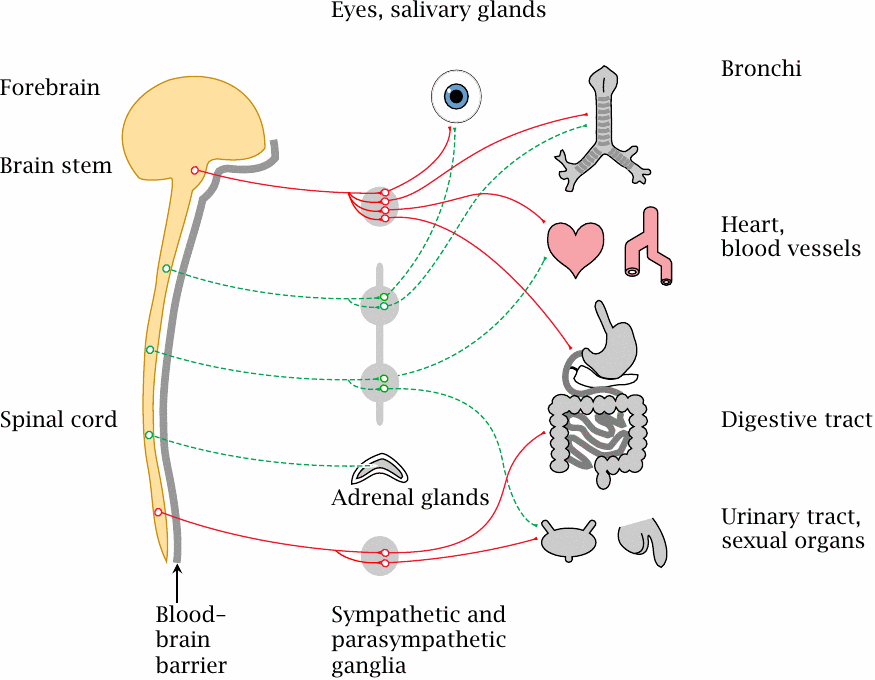
The autonomic nervous system comprises parts of both the central and the peripheral nervous system. It contains parasympathetic and sympathetic parts (red and green lines, respectively).
Within the autonomic nervous system, each connection between the CNS and a peripheral organ is made by two neurons connected in series. The cell body of the first neuron is located inside the CNS, but its efferent synapses are located inside a sympathetic or parasympathetic ganglion, outside of the blood brain barrier. The cell body of the corresponding second neuron is also located in this ganglion.
In most organs, the sympathetic and the parasympathetic nervous system exert antagonistic regulatory influences. The sympathetic nervous system will increase blood pressure and heart rate and will dilate the bronchi, while also diverting blood flow from the intestines and other interior organs toward the skeletal muscles. The parasympathetic nervous system will slow down cardiovascular function and promote perfusion and activity of the glands and digestive organs.
Most of the drugs covered in the following act on some type of synapse or other in the autonomic nervous system. The heart, the circulation, and the bronchi are particularly important as target organs for pharmacotherapy related to the autonomic nervous system.
| 6.13.2 |
Transmitter receptors in the peripheral autonomic nervous system |
| Subsystem | 1st Synapse | 2nd Synapse |
| sympathetic | nicotinic | α- or β-adrenergic |
| parasympathetic | nicotinic | muscarinic |
Nicotinic acetylcholine receptors are found in both the sympathetic and the parasympathetic ganglia. Inhibiting these receptors will therefore sweepingly inhibit both sympathetic and parasympathetic activity. On blood pressure, which is the single most important target parameter for drugs that act on the autonomic nervous system, the net effect will be a reduction. Nicotinic receptor antagonists can therefore be used to treat hypertension, and they were indeed so used in the past; however, because of their numerous side effects, they are now obsolete in this application.
Muscarinic acetylcholine receptors are found in the second synapses, that is, those between the second parasympathetic neurons and the effector cells such as gland and smooth muscle cells. Adrenergic receptors are found in the second synapses of the sympathetic nervous system. α-Adrenergic receptors are prominent in the blood vessels, β1 receptors in the heart, and β2 receptors in the bronchi and uterus. Drugs that act on these receptors have fewer side effects than nicotinic receptor agonists and antagonists, and they are more practically important in modern pharmacotherapy.
| 6.14 |
Pharmacology of adrenergic synapses |
The adrenergic synapses in the sympathetic nervous system are targeted by numerous drugs with multiple action modes. We will consider some representative examples.
| 6.14.1 |
Receptor agonists or antagonists and false transmitters at adrenergic synapses |
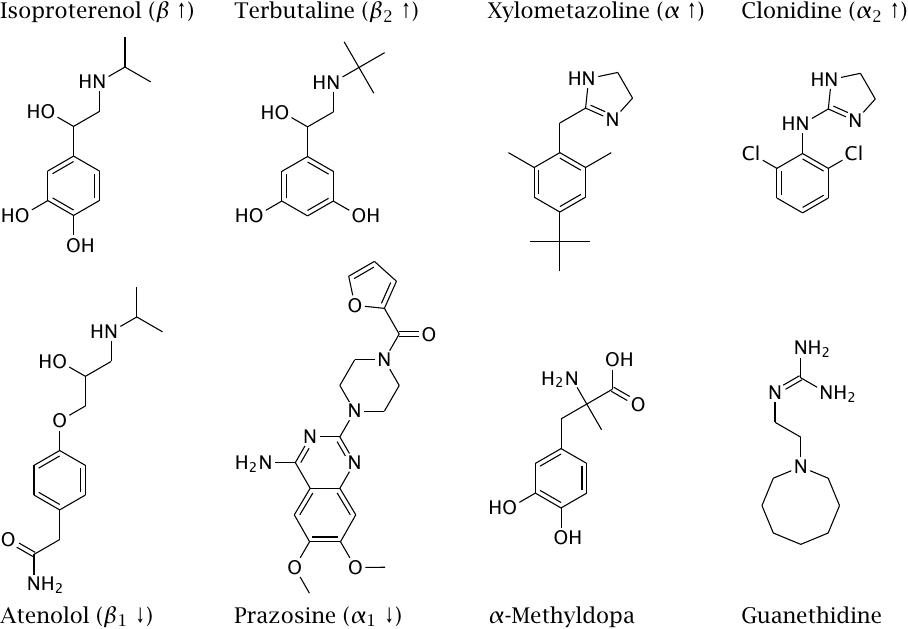
In this slide, up- and downward arrows indicate the activities of direct receptor agonists and antagonists, respectively.
Isoproterenol is used to increase the heart rate and the force of heart muscle contraction, while atenolol is used for the opposite purpose. β2-Selective adrenergic agonists like terbutaline relax smooth muscle in the bronchi and are used to treat asthma. They also relax smooth muscle in the uterus and are used to suppress premature labor.
Xylometazoline induces vasoconstriction and is applied locally to the nasal mucous membranes in order to suppress congestion. Clonidine was developed with similar intention, but turned out to be an α2-selective agonist [56]. Since α2 receptors prominently function in presynaptic feedback in adrenergic synapses, it actually reduces signaling efficiency in those synapses. It is used in the treatment of hypertension, as well as of various other conditions that range from pain to addiction and point to additional physiological roles of α2 receptors.
α-Methyldopa and guanethidine do not interact directly with adrenergic receptors; instead, they act as “false transmitters”. This peculiar mode of action is explained in the next slide.
| 6.14.2 |
Methyldopa is a false transmitter in noradrenergic synapses |
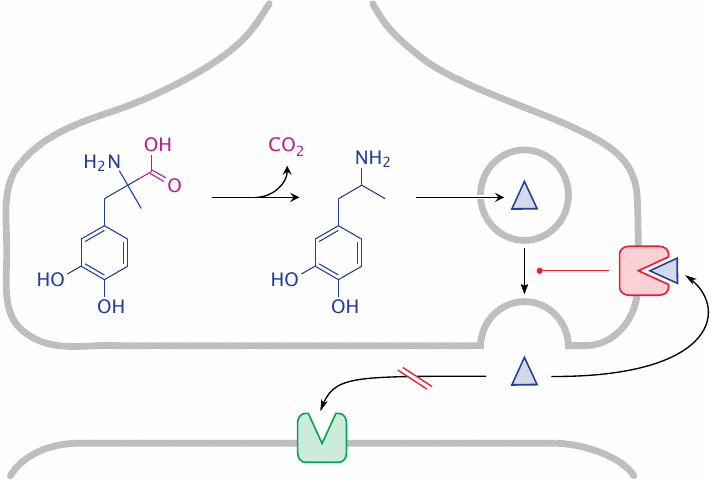
α-Methyldopa is a prodrug; after crossing the blood brain barrier in the same way as l-DOPA (see slide 3.5.5), it is taken up into adrenergic neurons and undergoes decarboxylation to α-methyldopamine. The metabolite is transported into presynaptic transmitter vesicles. The capacity of these vesicles for storing transmitter is limited, and therefore norepinephrine is displaced. When the presynaptic cell is activated, the false transmitter is released; it fails to stimulate postsynaptic α1 receptors but activates presynaptic α2 receptors, which exercise feedback inhibition on presynaptic transmitter release.
α-Methyldopa has been used for antihypertensive treatment but is no longer widely used. The drug guanethidine (shown in slide 6.14.1) acts in a manner similar to α-methyldopa, but without undergoing metabolic activation and only in the periphery, since it does not cross the blood brain barrier.
| 6.14.3 |
Drugs that act on the membrane transport of monoamine transmitters |
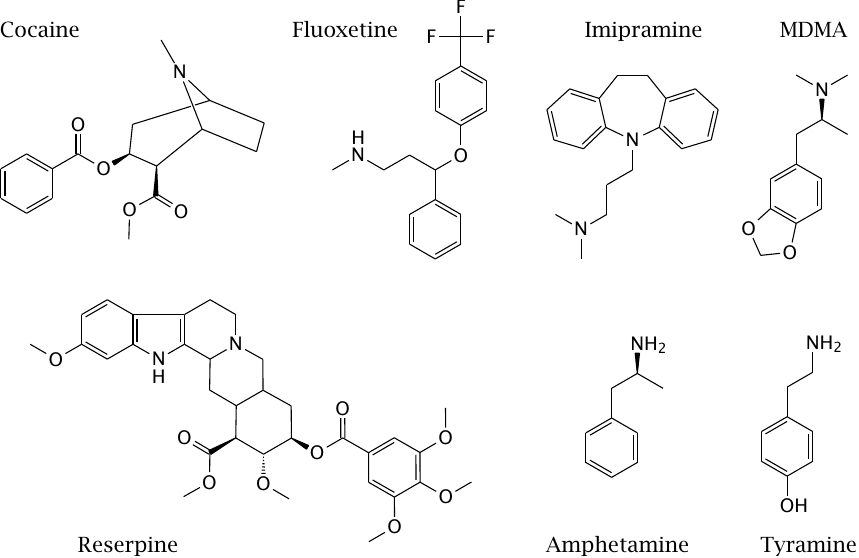
Aside from direct stimulation neurotransmitter receptors, the inhibition of presynaptic transmitter reuptake is another important mechanism by which synaptic signals can be amplified. This mechanism is important with psychoactive drugs of abuse as well as with regular antidepressant and antipsychotic drugs.
The reuptake transporters for the different monoamine transmitters are distinct but related; some drugs target specific transporters, while others have a broader specificity. Cocaine inhibits the presynaptic reuptake of dopamine, serotonin and norepinephrine, which accounts for its powerful stimulating effect.50
Fluoxetine and imipramine inhibit the presynaptic reuptake of serotonin and are used as antidepressants. The effect of fluoxetine is specific for serotonin, whereas imipramine also affects dopamine reuptake. In addition to these direct effects, enzymatic demethylation of imipramine gives rise to desipramine, which is also used as an antidepressant drug and blocks the reuptake of norepinephrine.
Amphetamine and tyramine release dopamine and norepinephrine from presynaptic storage vesicles and then promote their retrograde transport from the cytosol into the synaptic cleft. N-Methyl-3,4-methylenedioxymethamphetamine (MDMA, “ecstasy”) does the same to serotonin. It appears that the overall effect involves both specific interaction with the transporter proteins as well as non-ionic diffusion across cell and vesicle membranes [57].
Reserpine blocks the vesicular monoamine transporter (VMAT), which transports monoamine transmitters from the cytosol into storage vesicles in presynaptic cells. The VMAT is shared between adrenergic, dopaminergic, and serotoninergic synapses, and accordingly the effects of all of these transmitters are inhibited by reserpine. The drug has been used for antihypertensive therapy in the past, but it has been abandoned due to strong side effects, including impotence and depression.
Reserpine binds to its target noncovalently, but its affinity is so high that it will virtually never dissociate from its target, which illustrates that irreversible and covalent binding do not always coincide. It has recently attracted some renewed interest as an inhibitor of ABC transporters such as P-glycoprotein.51
| 6.14.4 |
Degradation of norepinephrine |
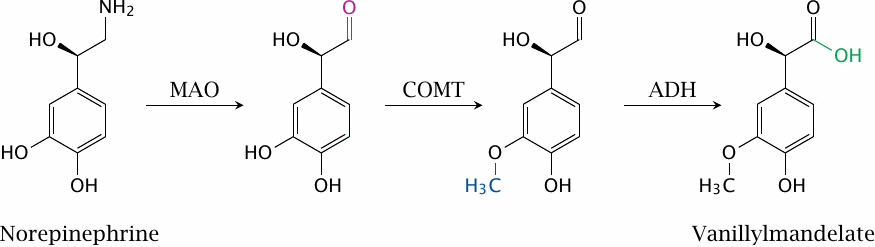
Synaptic signaling can also be influenced by inhibiting the degradation of transmitters. This slide illustrates the degradation pathway of norepinephrine; the pathways for dopamine and epinephrine are analogous. Either monoamine oxidase (MAO) or catechol-O-methyltransferase (COMT) may act on the substrate first, which gives rise to different intermediates but the same end product. The aldehyde group formed by monoamine oxidase can either be oxidized to the acid by aldehyde dehydrogenase (ADH) as shown here, or it can be reduced to the alcohol by aldehyde reductase. Both end products are excreted in the urine.
In the case of the catecholamines, both MAO and COMT can be targeted in order to increase synaptic transmitter levels. MAO is also involved in the degradation of serotonin. Monoamine oxidase inhibitors have been used in the past as antidepressants, but are now obsolete in this application. However, inhibitors of both MAO and COMT are still of interest as adjuvant treatments in Parkinson’s disease.
| 6.14.5 |
Reaction mechanism of monoamine oxidase (MAO) |
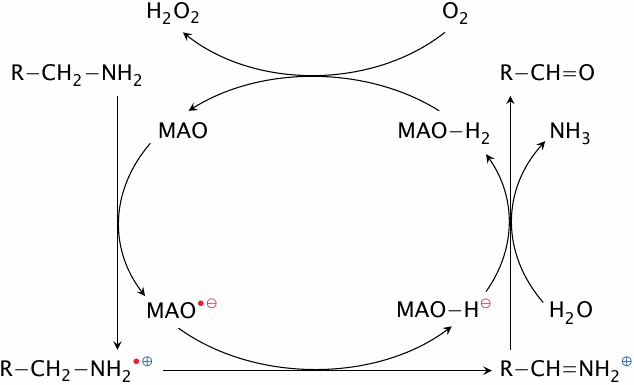
Monoamine oxidase contains a flavin coenzyme, which abstracts two electrons from the substrate in two successive steps. Abstraction of the first electron converts both the substrate and the enzyme to radicals. On the enzyme, the unpaired electron may remain with the flavin or migrate to a cysteine residue in the active site.
The abstraction of the second electron and of a proton converts the substrate to a Schiff base, which is then hydrolyzed to an aldehyde and free ammonia. MAO itself is reoxidized by molecular oxygen, which forms H2O2 in the process.
| 6.14.6 |
Mechanism-based inhibition of MAO by tranylcypromine |
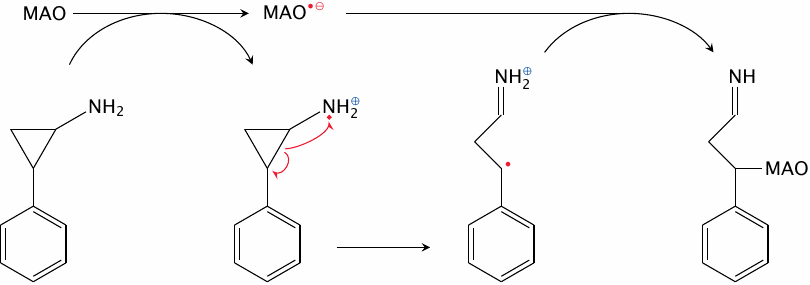
Tranylcypromine covalently inhibits monoamine oxidase by trapping the radical reaction intermediate. In the drug molecule, the amino group is attached to a reactive cyclopropyl ring, which upon the initial electron withdrawal opens up to produce a carbon radical. This carbon radical then combines with the radical intermediate of the enzyme.
MAO occurs not only in the brain but also in the liver. Its fairly broad specificity means that it can engage in some oxidative reactions in the metabolism of drugs and xenobiotics (see next slide). Liver MAO also scavenges aromatic amines such as tyramine, which are found in cheese and other fermented foods. Like amphetamine, tyramine releases norepinephrine and epinephrine from synaptic storage vesicles (see slide 6.14.3) and also from adrenal glands [58]. In patients treated with MAO inhibitors, the release of catecholamines by unscavenged tyramine can evoke an episode of hypertension, the so-called “cheese reaction.”
| 6.14.7 |
MAO-induced toxicity of MPTP (N-methyl-4-phenyl-tetrahydropyridine) |
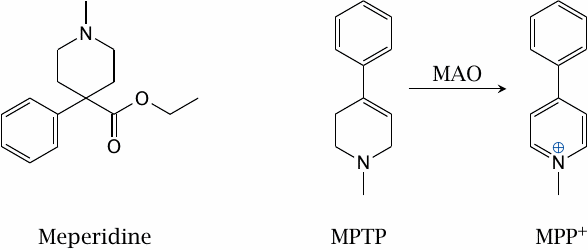
A xenobiotic metabolized by MAO is N-methyl-4-phenyl-tetrahydro-pyridine (MPTP). This is not a drug; instead, it has been found as a by-product in the synthesis of meperidine, a synthetic opioid. It enters dopaminergic neurons via the dopamine reuptake transporter. Inside the cells, it is converted by MAO to methyl-phenyl-pyridinium (MPP+), which inhibits complex I of the respiratory chain and causes cell death.
In the 1980s, a batch of MPTP-contaminated meperidine that had been made in an illicit lab in Vancouver caused several drug addicts in California to develop symptoms of Parkinson’s disease. MPTP has since been used to model Parkinson’s in experimental animals.
| 6.15 |
Pharmacology of cholinergic synapses |
As discussed above, acetylcholine has two major classes of receptors, which are selectively activated by muscarine and by nicotine, respectively. While nicotinic acetylcholine receptors are ionotropic, the muscarinic receptors are GPCRs. Muscarinic receptors occur in several subtypes, but most clinically used agonists or antagonists are not subtype-selective.
Both nicotinic and muscarinic receptors occur in the brain in various functions. In the periphery, nicotinic receptors occur in motor endplates and in autonomic ganglia, whereas muscarinic receptors occur in the second synapses of the parasympathetic nervous system. There are multiple clinical uses for drugs that target these receptors.
| 6.15.1 |
Structures of cholinergic receptor agonists |
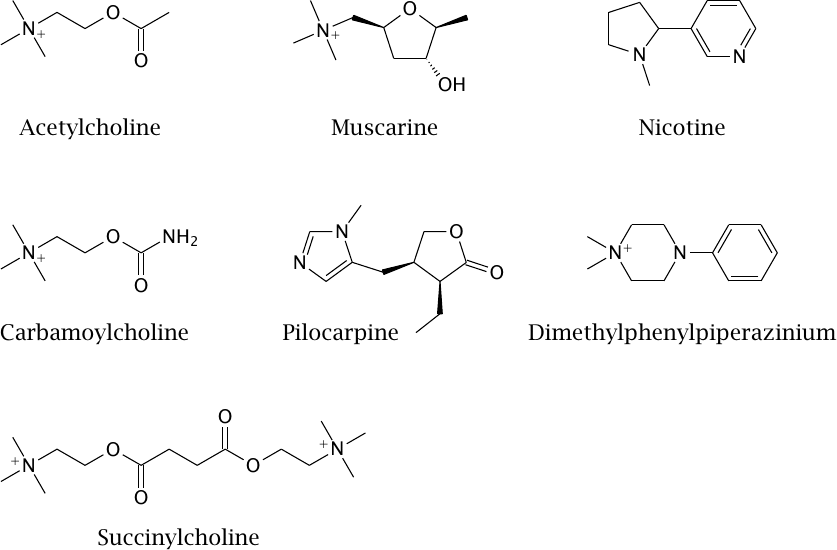
Like muscimol (slide 6.10.9), muscarine is found in fly agaric; it is not clinically used. Pilocarpine is a muscarinic agonist that is used in ophthalmology to promote the discharge of fluid from the eyeball in order to reduce intraocular pressure in glaucoma patients.52 Carbamoylcholine is active on both muscarinic and nicotinic receptors, but more so on the former. It is used occasionally following abdominal surgery for making a drowsy, hung-over intestine sit up and take notice.
The stimulating effect of nicotine is mediated by nicotinic receptors in the brain. In the periphery, the drug stimulates the first synapses in both the sympathetic and the parasympathetic nervous system (see slide 6.13.1); apart from increased blood pressure, consequences also include accelerated bowel movements, which in former, less pedagogically wholesome times could be nicely observed in small boys partaking of their grandfathers’ cigars. Dimethylphenylpiperazinium is a nicotinic agonist that is excluded by the blood brain barrier and is useful in physiological experiments.
As stated earlier, the nicotinic acetylcholine receptors in the motor endplates differ in their subunit composition from those in the autonomic ganglia. Succinylcholine is a selective agonist for the subtype found in motor endplates. By a mechanism that involves receptor desensitization (see slide 6.10.6), it renders the muscle cells unresponsive to presynaptically released acetylcholine; the effect is muscle relaxation. This is used as an adjuvant to systemic narcosis. Succinylcholine is cleaved and inactivated within minutes by a cholinesterase in blood plasma.
| 6.15.2 |
Structures of cholinergic receptor antagonists |
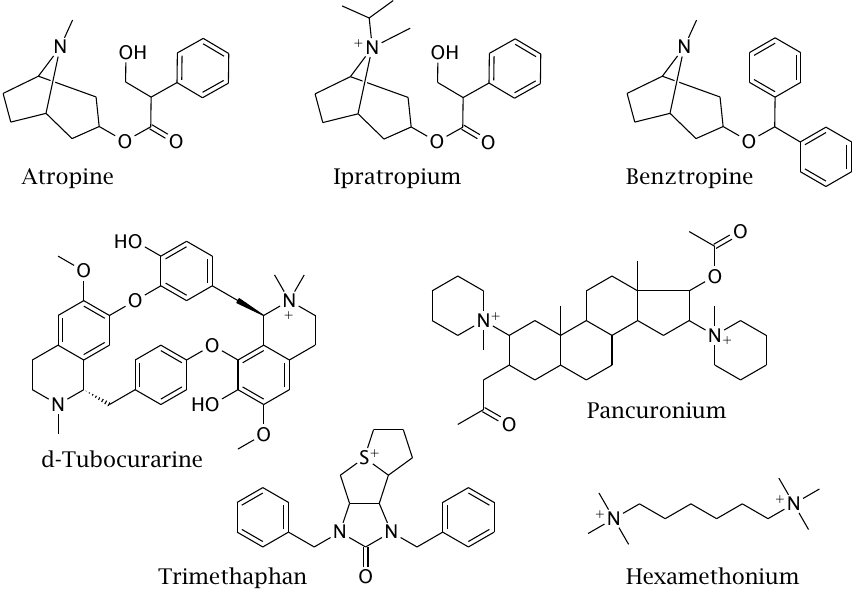
Atropine, ipratropium bromide and benztropine are all muscarinic antagonists. Atropine is used to suppress bronchial secretion in preparation of intubation for inhalation narcosis. Ipratropium is excluded by the blood brain barrier and therefore more suitable than atropine for sustained treatment; it is used to relax bronchi in asthma patients and, with cardiac patients, to set a little fire to the pants of a sluggish sinoatrial node.
In contrast to ipratropium, benztropine has been designed for increased penetration of the blood brain barrier. It targets muscarinic synapses in the brain that are functionally antagonistic to dopaminergic ones and is used as an adjuvant treatment in Parkinson’s disease.
Pancuronium and d-tubocurarine are antagonists at nicotinic receptors in motor endplates. While their effect on the receptor is opposite to that of succinylcholine, the end result is the same—neuromuscular blockade and muscle relaxation, and they are used for the same purpose.53
Trimethaphan and hexamethonium are “ganglion blockers,” that is, antagonists selective for the nicotinic receptor subtype found in autonomic ganglia. They have powerful antihypertensive effects but are no longer commonly used in that application.54
| 6.15.3 |
The catalytic mechanism of cholinesterase |
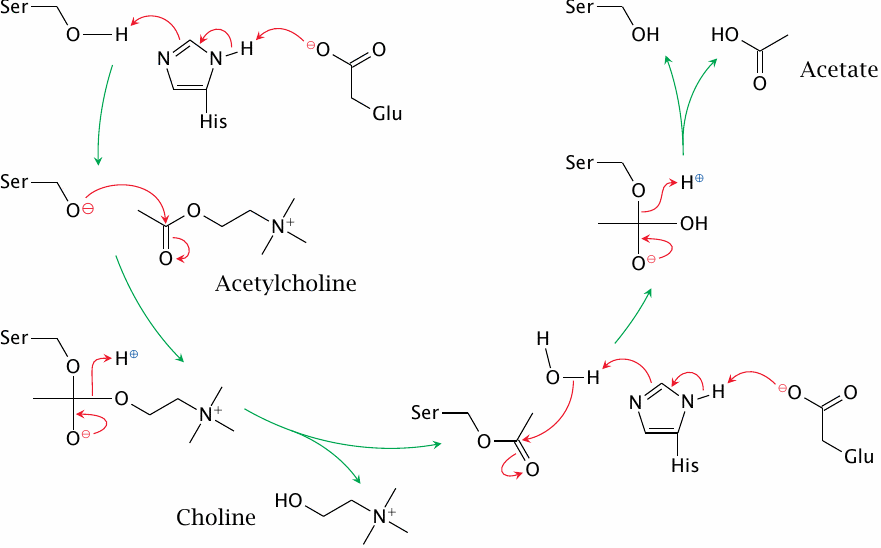
In both nicotinergic and muscarinergic synapses, acetylcholine is inactivated by cholinesterase; inhibitors of the enzyme will thus affect both types of synapses. They are of limited use in medicine but are more widely used as poisons.
The catalytic mechanism of cholinesterase resembles that of serine proteases. In the active site, a glutamate and a histidine residue cooperate in deprotonating the catalytic serine, which then performs nucleophilic attack on the substrate. This results in a tetrahedral transition state, which rapidly converts to an acylated form of the enzyme; the latter is then hydrolysed. Cholinesterase is very efficient; because of this, the chance of any single acetylcholine molecule binding to a receptor more than once is negligible.
| 6.15.4 |
Covalent inactivation of cholinesterase by DFP, and its reactivation by pralidoxime |
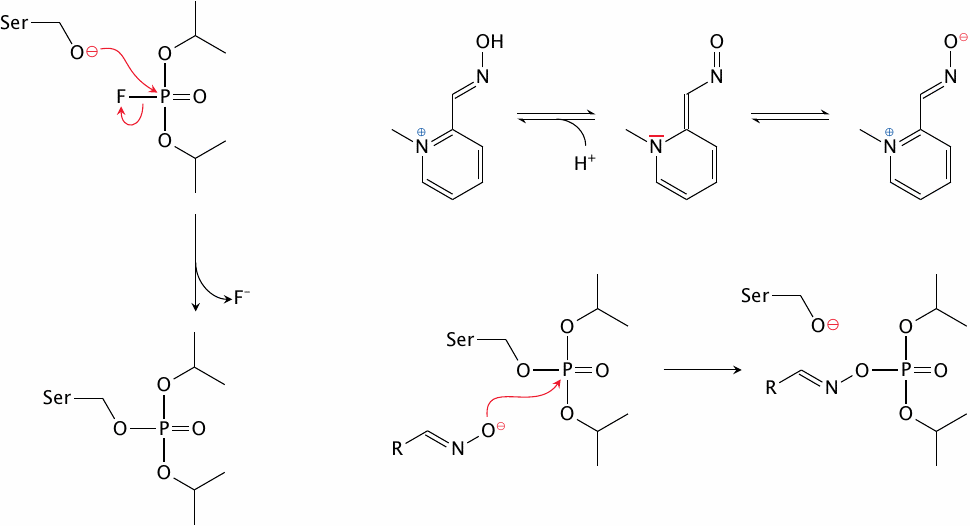
Organophosphate inhibitors such as diisopropylfluorophosphate (DFP) are sterically similar to the tetrahedral transition state of the cholinesterase reaction and therefore bind the enzyme very avidly, before reacting with it covalently (top). The modification can be reversed with oximes (bottom) such as pralidoxime (see slide 6.15.5).
DFP and several structurally similar compounds are extremely toxic to humans; they are taken up by inhalation and also penetrate intact skin. They were mass-produced for chemical warfare by all major parties in World War II but never used in that war. However, Iraq’s Saddam Hussein used them in his war on Iran in the 1980s.
| 6.15.5 |
Structures of cholinesterase inhibitors and of a reactivator |
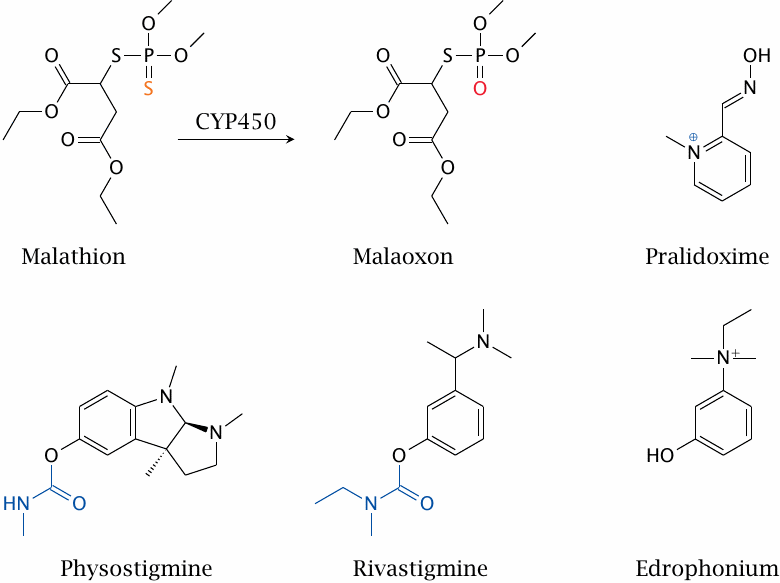
Organophosphates are also widely used as insecticides. In malathion, the fluoride leaving group found in DFP has been replaced by a substituted thiol. In addition, one of the doubly bonded oxygens has also been replaced by sulfur. Before reacting with cholinesterase, this second sulfur atom has to be replaced with oxygen by cytochrome P450, which yields malaoxon. The sulfur-substituted precursor form is deployed because it is more environmentally stable.
The two ester groups in the side chains of the leaving group constitute a safety feature. They are susceptible to cleavage by esterases in human blood plasma. Esterase cleavage competes with activation by cytochrome P450, and the cleavage product no longer reacts with cholinesterase. Malathion is thus much less toxic to humans than other, older organophosphates.
Edrophonium is a noncovalent inhibitor, whereas rivastigmine and physostigmine are covalent ones; the groups that end up bound to the active site serine are highlighted in both. Pralidoxime is a cholinesterase reactivator that is used in the treatment of organophosphate poisonings.
Clinical uses of cholinesterase inhibitors concern the reduction of intraocular pressure in glaucoma (see the footnote to section 6.15.1), and the amplification of cholinergic signaling in motor endplates, which is used in myasthenia gravis. In the latter disease, nicotinic acetylcholine receptors in motor endplates are inactivated by autoantibodies, which leads to muscle weakness; extending the lifetime of acetylcholine can improve the compromised synaptic function.
| 6.16 |
Drugs that interact with purine or opioid receptors |
There are many more neurotransmitters; we will mention just two more classes that have considerable importance in pharmacology.
| 6.16.1 |
Purine receptor agonists and antagonists |
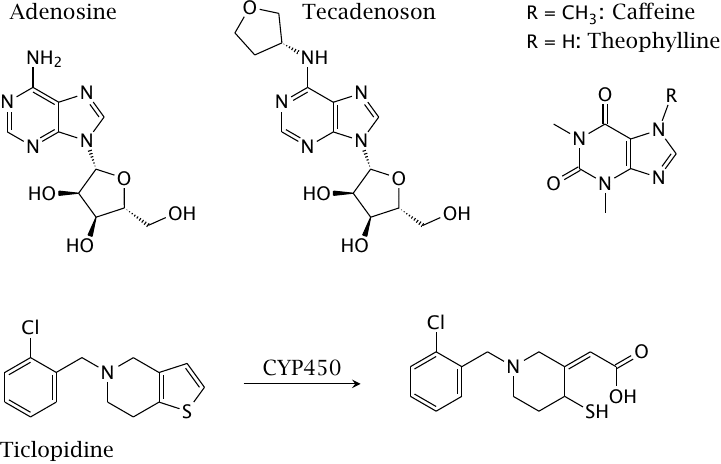
Purine receptors fall into two structural families. The P2X receptors constitute an independent family of ligand-gated channels; all others belong to the GPCR family. The receptors serve in different roles inside and outside the nervous system.
Adenosine receptors are often found alongside catecholamine receptors in the same synapses, where they tend to counteract the effects of the latter. Accordingly, the effect of inhibiting these adenosine receptors resembles that of stimulating the catecholamine receptors, and vice versa. As an aside, adenosine receptors have also been reported to form hetero-oligomers with GPCRs specific for various other neurotransmitters. Caffeine and theophylline are A2A adenosine receptor antagonists that are used as stimulants. Theophylline is also effective in the treatment of asthma. Tecadenoson is an adenosine receptor agonist that is used in the treatment of some forms of cardiac arrhythmia. Ticlopidine blocks ADP receptors on thrombocytes, which inhibits their aggregation; this is useful in the treatment of atherosclerosis (see slide 10.6.7). It requires metabolic activation by cytochrome P450.
| 6.16.2 |
Opioid receptor agonists and antagonists |
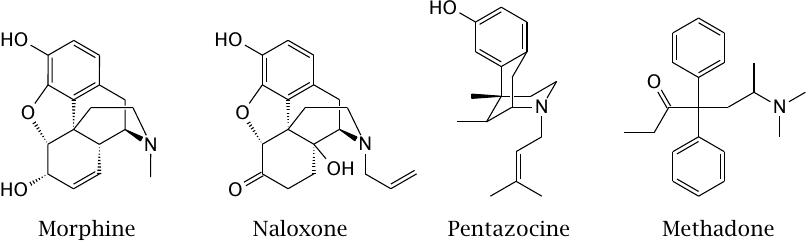
Opioid receptors are very effective at suppressing pain perception; their endogenous agonists are endorphins. Morphine is an agonist at µ, κ and δ-opioid receptors. Naloxone is an antagonist at the same receptors and is used in opioid poisonings. Pentazocine acts as a partial agonist primarily at µ-opioid receptors; it is less potent than morphine yet still a strong analgesic. Methadone is a full µ-receptor agonist and is used as a legal substitute for heroin; more recently, a combination of pentazocine and naloxone has come into use as a milder alternative to methadone.
We had previously considered the formation of κδ-receptor heterodimers and their selective activation by 6-guanidinonaltrindole (see slide 5.6.3).