| 3 |
Pharmacokinetics |
| 3.1 |
The scope of pharmacokinetics |
Pharmacokinetics deals with the following questions:
- Will the drug reach its intended site of action? If not, can we improve the drug’s uptake and distribution to help it reach its target?
- After uptake, how long will the drug stay in the system? How is it eliminated from the system?
As we will see, getting a drug to its target can be a considerable challenge. Many experimental drugs that are viable in principle and which can be shown to work in vitro are not useful in vivo because of this difficulty.
And no, the latest and greatest nanoparticles aren’t going to be the answer to this problem. You may have noticed that most people who flog their revolutionary nanoparticle contraptions for “drug delivery” don’t even talk about any specific drugs that they are going to deliver, or to what location exactly. This should tell us something.
| 3.2 |
Stages of drug transport |
- Absorption: Uptake of the drug from the compartment of application into the blood plasma
- Distribution: Equilibration of the drug between the blood plasma and the rest of the organism
- Elimination: Excretion or metabolic inactivation of the drug
These stages do not occur strictly successively but overlap in time: As soon as some drug molecules have been taken up, they will start to distribute and undergo elimination, even while most others are still waiting for uptake. Moreover, strictly speaking, equilibrium of distribution can be reached only when a drug is administered continuously, e.g. by intravenous infusion, which is not usually the case. With these caveats out of the way, we will now consider these stages separately and in turn.
| 3.2.1 |
Absorption depends on the route of application |
| Route | Advantage | Disadvantage |
| Oral | Convenience—route of choice where possible | Multiple barriers between intestine and circulation |
| Intravenous | No barriers to absorption | Involved; risk of infection; allergic reactions more severe |
| Pulmonary | Fast, quantitative uptake | Limited to gases (mostly narcotics) |
Oral application of drugs is the most common case, because it is the most convenient for the patients. However, the obstacles to uptake from the gut into the circulation are formidable; we have already seen examples of drugs—remikiren and saralasin, see slide 1.2.6f—that fail entirely to be absorbed after oral application.
The second-most common route of application is intravenous injection or infusion. In this case, the absorption stage is bypassed altogether. This route is used with most protein drugs, as well as with small molecules that fail to be taken up after oral ingestion.
In clinical emergencies, intravenous application is usually preferred even with drugs that are suitable for oral application in principle, in order to ensure their rapid and quantitative uptake.
| 3.2.2 |
The need for distribution varies with the location of the drug target |
| Location of target | Example |
| Inside circulation, outside cells | Proteases in blood coagulation and fibrinolysis |
| Inside circulation, inside cells | Chemotherapy of malaria parasites |
| Outside circulation, on cell surfaces | Histamine receptors |
| Outside circulation, inside cells | Cyclooxygenase |
After reaching the circulation, a drug may have to cross fewer or more barriers along the way to its target. In the most favorable case, the targets are located right within the circulation, and not hidden within any cells. This applies to the proteases involved in blood clot formation and dissolution (fibrinolysis), as well as to receptors that are located on the surfaces of thrombocytes or leukocytes.
Targets are less accessible when they are placed outside the circulation, yet less so when placed within cells, and least when placed inside the central nervous system. We will now consider the anatomical reasons for this order of precedence.
| 3.3 |
Drug transport across anatomical barriers |
Anatomical barriers concern all stages of drug transport. They differ in their active and passive transport properties. Because of its central place in drug transport, the blood circulation and its anatomical properties are particularly pertinent.
| 3.3.1 |
Sections of the blood circulation |
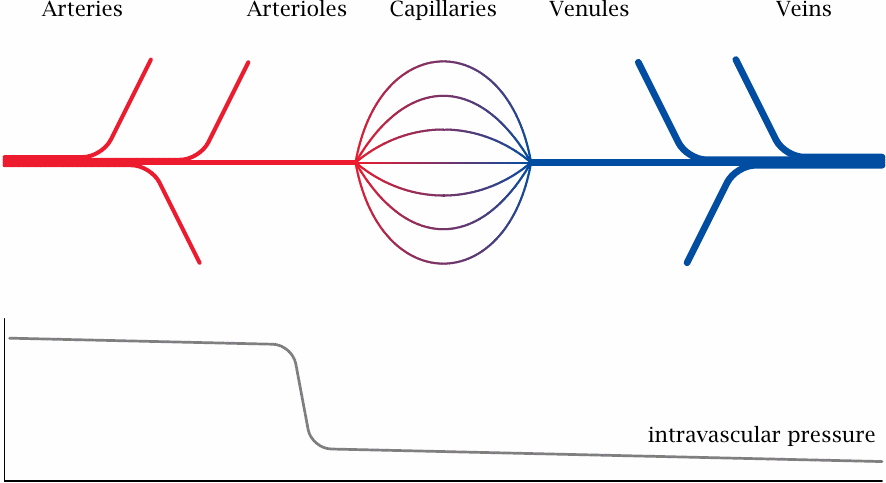
In the general circulation, oxygen-rich blood flows in the arteries. The final branches of the arteries are the arterioles. These vessels have the greatest flow resistance, and therefore the intravascular blood pressure, which is high in the arteries, here drops steeply to the much lower level that prevails in the capillaries and veins.
Both the arteries and veins have walls with multiple continuous layers of tissue that have low permeability for gases and metabolites. The exchange of oxygen, carbon dioxide, and other solutes between blood and tissues is mostly confined to the capillaries.
| 3.3.2 |
Capillaries as barriers to drug transport |
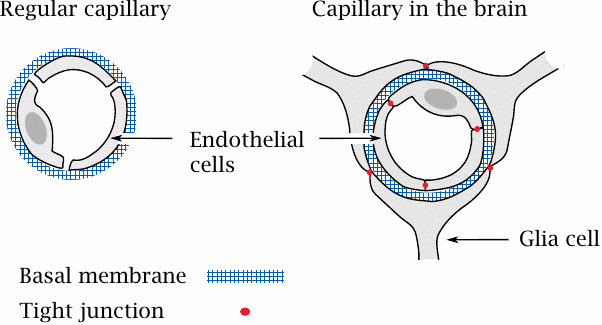
Capillaries are the ubiquitous, tiny blood vessels—just wide enough to permit passage of blood cells in a single file—that mediate the exchange of oxygen, CO2 and metabolites between the blood within and the tissue without. Unlike the multi-layered arteries and veins, a capillary has just a single cell layer, the endothelium, which is surrounded by an acellular meshwork composed of glycans and proteins, the basal membrane.
In almost all tissues outside the brain, the capillary walls have a loose, leaky structure, such that there are gaps between, and fenestrations (windows) across the endothelial cells. The only continuous structure that spans these gaps and fenestrations is the basal membrane, which therefore forms the only barrier to substance exchange by diffusion. It acts like a dialysis membrane: molecules smaller than 10 kDa can pass it freely, whereas blood plasma proteins and other macromolecules are retained.
In the brain and the spinal cord, the endothelia of the capillaries don’t have fenestrations, and adjacent cells are connected by tight junctions. On the outer side, the basal membrane of each capillary is surrounded by another tightly sealed cellular layer, which is formed by glia cells. Therefore, instead of a single, porous basal membrane, the lumen of the capillary is separated from the surrounding tissue by two continuous cell layers. This structure, which is much less permissive toward substance exchange than an ordinary capillary wall, forms the blood brain barrier.
The stacked cell membranes of the blood brain barrier prevent many small molecules from entering the central nervous system. In addition to passively impeding diffusion, the endothelial cells also contain ABC transporters that actively eject many drug molecules from the cytosol right back into the circulation. These transporters further reduce the penetration of drugs into the brain (see slide 3.5.3).
| 3.3.3 |
The intestinal epithelium as a barrier to drug absorption |
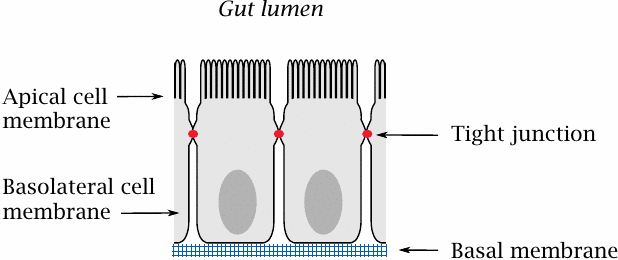
The small intestine is the place in which most orally ingested drugs are taken up. The epithelial cells sit atop a basal membrane and are joined to each other by tight junctions; therefore, drug molecules (and nutrients) have to be taken up across the cell membranes.
Like the endothelial cells in the blood brain barrier, the intestinal epithelia express ABC transporters, which extrude some solutes from the cells back into the gut lumen. Moreover, these cells also express cytochrome P450 enzymes, which metabolize and inactivate many drug molecules. The role of cytochrome P450 enzymes in drug metabolism is covered in detail in Chapter 4.
| 3.4 |
Drug transport across cell membranes |
As we have seen, cell membranes restrict drug transport across the intestine and into the brain. In addition, they will also restrict the cellular uptake of drugs that act on intracellular targets.
| 3.4.1 |
Mechanisms of solute transport across cell membranes |
-
Active transport
- Primary: ATP-coupled
- Secondary: driven by ion gradients
-
Passive transport
- Facilitated diffusion: protein-mediated transport, not coupled to ATP or ion gradients
- Non-facilitated diffusion of lipophilic compounds; non-ionic diffusion
Transport of types 1a, 1b, and 2a is mediated by transport proteins and is substrate-specific. Some drugs can take advantage of active transport; an example is cytosine arabinoside, which is taken up from the gut by the same transporter that also mediates uptake of cytidine and deoxycytidine (see slide 12.6.7). However, most drugs don’t fit the substrate specificities of any transport protein that would aid in uptake and distribution. Therefore, they have to cross cell membranes by non-facilitated diffusion, which occurs directly across the lipid bilayer and is independent of membrane proteins. The efficiency of this mode of transport depends mostly on the drug’s physicochemical properties.
| 3.4.2 |
Structure of a phosphatidylcholine bilayer |
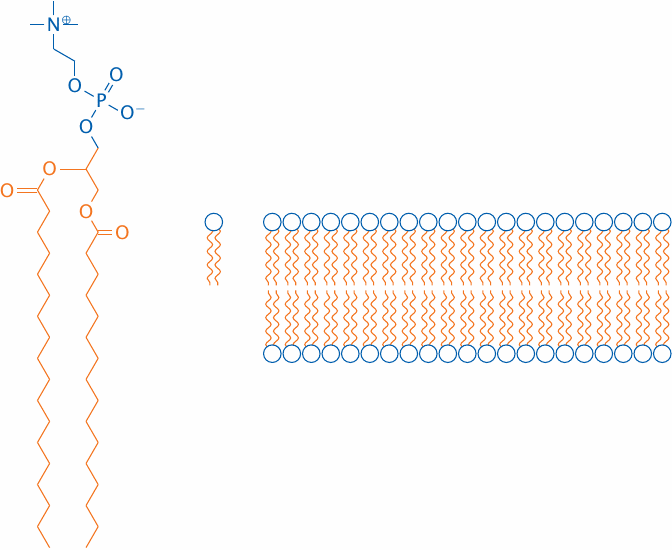
Obviously, cell membranes are more complex than a simple phosphatidylcholine (PC) bilayer; about 50% of a cell membrane consists of various membrane proteins, and the remainder is composed of a fairly complex mixture of lipid molecules. Nevertheless, a simple PC membrane is a useful model for drug transport across cell membranes by passive diffusion.
The solvent interfaces of a PC bilayer consist of the hydrophilic glycerophosphocholine headgroups, whereas the core consists of the aggregated fatty acyl side chains.
| 3.4.3 |
The polarity of drug molecules affects their rate of diffusion across lipid bilayers |
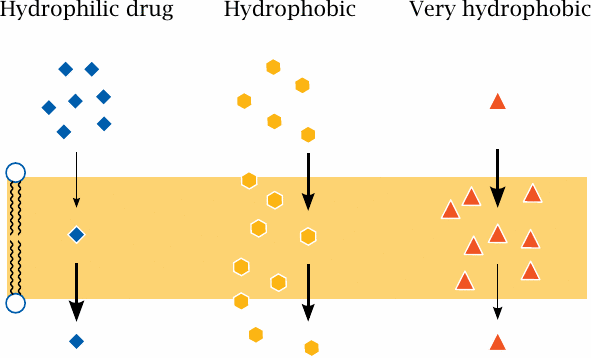
The ability of drug molecules to cross a lipid bilayer by passive diffusion correlates with their ability to partition into and out of the hydrophobic membrane interior. A fairly but not excessively hydrophobic character is most conducive to transport; very polar molecules will fail to enter the membrane, whereas extremely hydrophobic molecules will enter readily but may have a hard time leaving it on the other side.
| 3.4.4 |
The membrane permeability of drugs can be improved by removing polar functional groups |
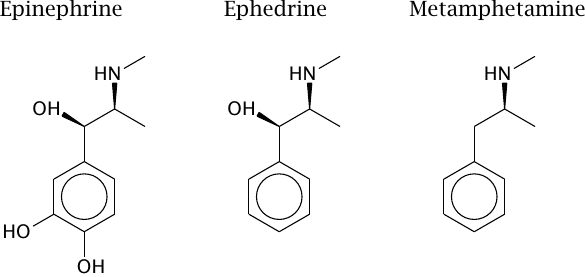
Removal of charged and polar functional groups can improve the ability of a drug molecule to cross lipid bilayers. For example, ephedrine and metamphetamine are analogues of epinephrine that have shed some hydroxyl groups. Accordingly, they penetrate cell membranes more readily than the parent compound, which promotes their uptake from the intestine and their distribution to the brain.
However, the structural changes also cause a shift in the molecular mode of action. Like epinephrine, both ephedrine and metamphetamine stimulate adrenergic synapses; however, while epinephrine acts directly on adrenergic receptors at the cell surface, ephedrine and metamphetamine act primarily on intracellular targets (see section 6.14.3). This example illustrates that the scope of structural alterations for the sake of improving pharmacokinetic properties is limited.
| 3.4.5 |
Resorption esters can improve the diffusion of drugs across membranes |
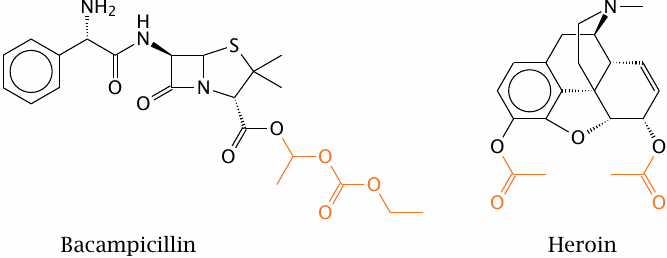
An alternate approach to improving the membrane penetration of drug molecules is to turn them into prodrugs, in which polar or ionizable groups are masked with hydrophobic residues. After uptake, these hydrophobic groups must be able to undergo enzymatic cleavage in order to release the original drug molecules. Most commonly, such functional groups are organic esters.
Bacampicillin is such a resorption ester; it contains ampicillin, which after uptake into the intestinal epithelium is released by esterases. Heroin is morphine with two acetyl groups added so as to mask two polar hydroxyl groups and facilitate distribution of the drug to the central nervous system. Once there, heroin undergoes cleavage by esterases, which releases the morphine again. In both prodrug molecules, the cleavable ester groups are highlighted.
| 3.4.6 |
History of heroin |

Heroin is a semisynthetic drug, which is obtained from morphine through acetylation.17 It was originally intended not as a narcotic drug but rather as an analgesic milder than morphine that would be suitable for everyday use; and as these pictures illustrate, it was indeed marketed for a while in this vein. However, because of its improved penetration into the brain, it is actually stronger and more addictive than morphine.
| 3.4.7 |
Ionizable drug molecules may cross bilayers by non-ionic diffusion |
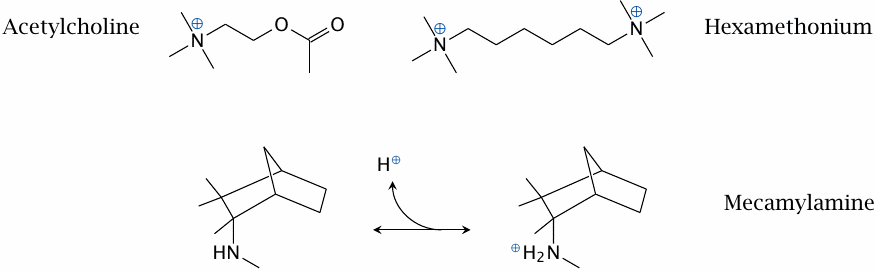
The physiological mediator acetylcholine possesses a quaternary amino group whose positive charge is essential for the interaction with the nicotinic acetylcholine receptor (NAR; see slide 6.10.1).
Hexamethonium is an inhibitor of the NAR. Since it contains two quaternary amino groups, it is transported across membranes very poorly and cannot be orally applied. In contrast, the inhibitor mecamylamine contains a tertiary amine. Therefore, it can cross bilayers in its unprotonated form and then bind a proton to acquire the charge necessary for receptor binding. This drug is amenable to uptake after oral ingestion. The transport across membranes of an ionizable drug in its non-ionized form is called non-ionic diffusion.
| 3.4.8 |
Gastric acid promotes accumulation of acetylsalicylic acid in the cells of the mucous membrane |
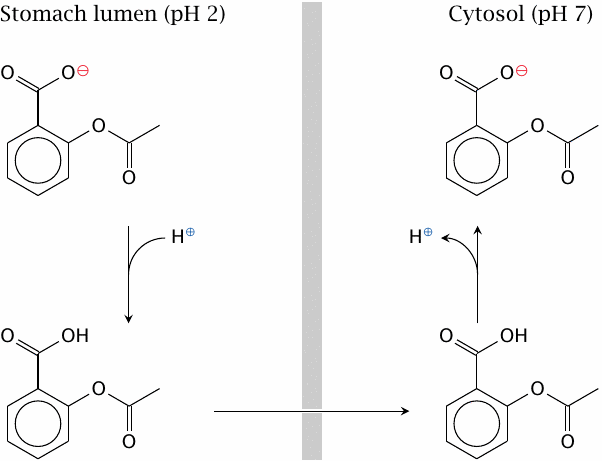
Non-ionic diffusion will be affected by pH gradients. The steepest such gradient exists at the surface of the gastric mucous membrane. The high proton concentration inside the stomach lumen can cause accumulation of acetylsalicylic acid inside the gastric epithelial cells and promote gastritis and ulcer formation.
To prevent the accumulation of acetylsalicylic acid in the gastric mucosal cells, the drug can be given an enteric coating, that is, be encapsulated in some sort of polymer that resists dissolution at the low pH in the stomach but readily dissolves in the slightly alkaline milieu of the small intestine (see slide 14.4.1).
| 3.5 |
Protein-mediated drug transport |
Non-specific, passive diffusion is the most common mode of drug transport across cell membranes; nevertheless, many drugs, and in particular drug metabolites, are also subject to protein-mediated active or passive transport. An important class of membrane proteins involved in drug transport are the so-called ABC transporters. The “ABC” in this name stands for “ATP-binding cassette,” which denotes a structural motif that is conserved among these proteins. ABC transporters are found in many places, but prominently in the small intestine, the liver, the kidneys, and the blood brain barrier. Typically, they oppose the intracellular accumulation of drug molecules by extruding them back out of the cell, for example into the gut lumen or into the blood stream.
ABC transporters tend to have rather broad substrate specificities, and they can therefore transport/extrude a rather large number of drugs. Overexpression of ABC transporters in tumor cells is an important mechanism of resistance to anticancer drugs. Similarly, ABC transporters in bacteria can cause resistance to antibiotics.
Organic cation transporters and organic anion transporters are other major types of membrane proteins involved in drug transport, particularly in the kidneys and the liver. They are not driven by ATP, but some are powered by ion co-transport or antiport; that is, they perform secondary active transport. Like ABC transporters, organic anion and cation transporters tend to have broad substrate specificities.
| 3.5.1 |
Inward- and outward-facing conformations of ABC transporters |
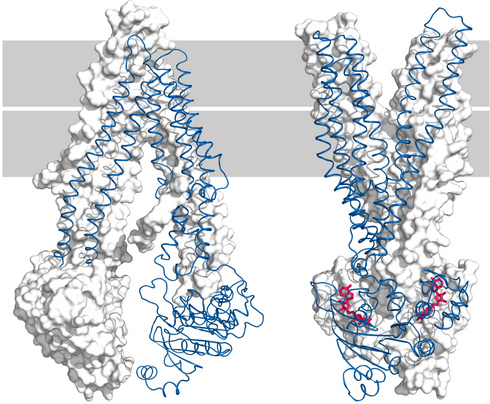
Bacterial and mammalian ABC transporters share a high degree of structural homology. The left panel of this slide shows the structure of P-glycoprotein in its inward-facing conformation [12]). P-glycoprotein—also called MDR-1, for multi-drug resistance—is a human ABC transporter notorious for its role in the resistance of tumor cells to anticancer drugs.
The right panel shows the outward-facing conformation of the bacterial ABC transporter Sav1866 [13]. In each panel, one of the two homologous functional domains (MDR-1) or separate monomers (Sav1866) is rendered in cartoon mode and in blue, while the other is shown in white and in surface representation.
The Sav1866 structure also contains two molecules of a non-hydrolyzable ATP analogue—shown in red—within the ATP binding sites, which are located at the interface of the two subunits.
| 3.5.2 |
The functional cycle of ABC transporters |
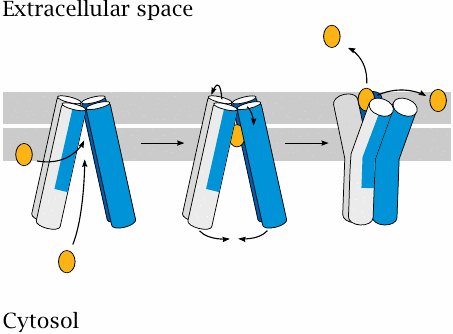
The ABC transporters alternate between the inward- and the outward-facing conformation; the cyclical transition between them is powered by ATP hydrolysis.
ABC transporters are involved in the transport of both drugs or other solutes and of membrane lipids. Accordingly, substrates may be accepted from the cytosol or from the inner leaflet of the cell membrane. The ATP-powered conformational change first sequesters the cargo molecule inside the transporter and then expels it into the outer membrane leaflet or the extracellular space.
| 3.5.3 |
ABC transporters and the blood brain barrier (1) |
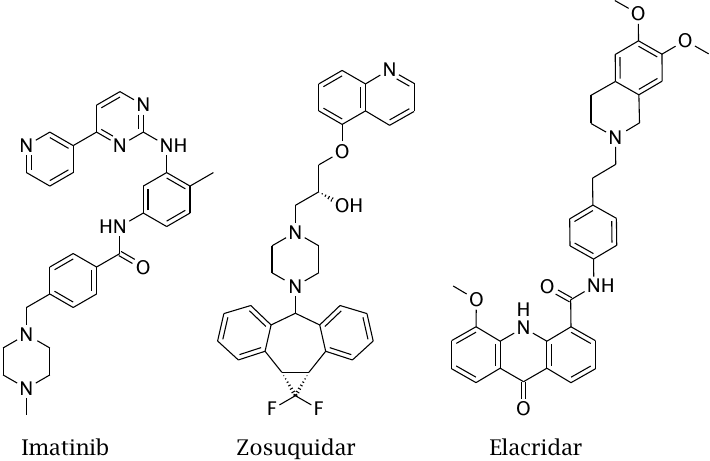
Imatinib is an anticancer drug. Its penetration into the brain across the blood brain barrier is inhibited by ABC transporters, which capture the drug inside the endothelial cells of the capillaries and extrude it back into the circulation.
Zosuquidar and elacridar are ABC transporter inhibitors. They have been developed primarily to overcome ABC transporter-mediated drug resistance in cancers. However, they also inhibit ABC transporters in normal cells.
| 3.5.4 |
ABC transporters and the blood brain barrier (2) |
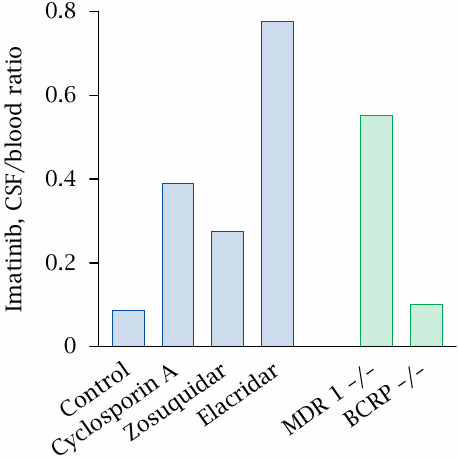
This experiment illustrates the effect of ABC transporters on the penetration of imatinib through the blood brain barrier. The concentration of imatinib was measured both in the cerebrospinal fluid (CSF) and the blood. A low ratio of CSF to blood concentration indicates a low degree of brain penetration.
When imatinib is applied alone, its concentration in the CSF is only 10% of that in the blood. Penetration of imatinib increases when it is applied together with cyclosporin A—an immunosuppressive drug, which inhibits ABC transporters as a side effect—or with zosuquidar, both of which are known inhibitors of MDR-1. An even stronger effect is observed with elacridar, which inhibits both MDR-1 as well as BCRP, another ABC transporter involved in cancer cell resistance (BCRP stands for “breast cancer resistance protein”).
In order to identify the transporters whose inhibition caused the increased brain penetration of imatinib, the drug effects were compared with genetic knock-out of the transport proteins. The effect of knocking out MDR-1 (MDR-1 –/–) is strong enough to account for the action of zosuquidar and cyclosporin A, but it falls short of the inhibition observed with elacridar. Surprisingly, however, a genetic knock-out of BCRP (BCRP –/–) has hardly any effect on imatinib penetration. This suggests that transporters other than MDR-1 and BCRP participate in imatinib extrusion and are inhibited by elacridar. Figure prepared from original data in [14].
Inhibition of ABC transporters also occurs as a side effect of other, more commonly prescribed drugs. In a clinical case report [15], the inhibition of ABC transporters by verapamil, a calcium channel blocker which had been prescribed for a heart ailment, occasioned CNS penetration and toxicity by colchicine, which had been prescribed simultaneously for gout.
| 3.5.5 |
l-DOPA reaches the brain by specific transport |
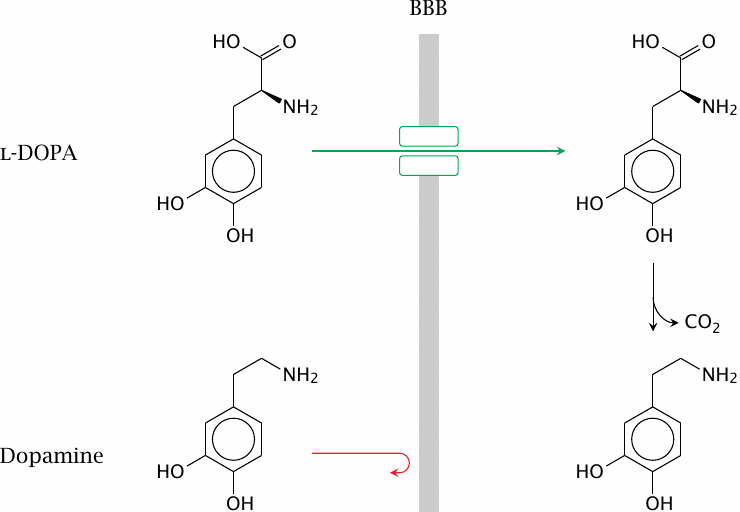
Occasionally, active transport can work in favor of drug distribution. An important example is l-DOPA, which is the biosynthetic precursor of the neurotransmitter dopamine. In Parkinson’s disease, a specific group of dopamine-producing cells in the brain stem—named the substantia nigra—degenerates, and the ensuing lack of dopamine causes the symptoms of the disease.
Parkinson’s disease can be treated (not cured) by the replacement of dopamine. However, dopamine itself is not able to cross the blood brain barrier; we therefore need a prodrug. Fortunately, the immediate precursor of dopamine, l-DOPA, structurally resembles the aromatic amino acid tyrosine, and it is transported across the blood brain barrier by the same transporter as is tyrosine.
Once inside the brain, l-DOPA can undergo enzymatic decarboxylation to dopamine. Application of l-DOPA is an important element in the treatment of Parkinson’s disease.18
| 3.6 |
Anatomy and physiology of drug uptake and distribution |
In addition to the general structures of endothelial and epithelial cell layers, some aspects of organ anatomy and physiology are important in controlling the uptake and distribution of drugs.
| 3.6.1 |
The portal circulation |
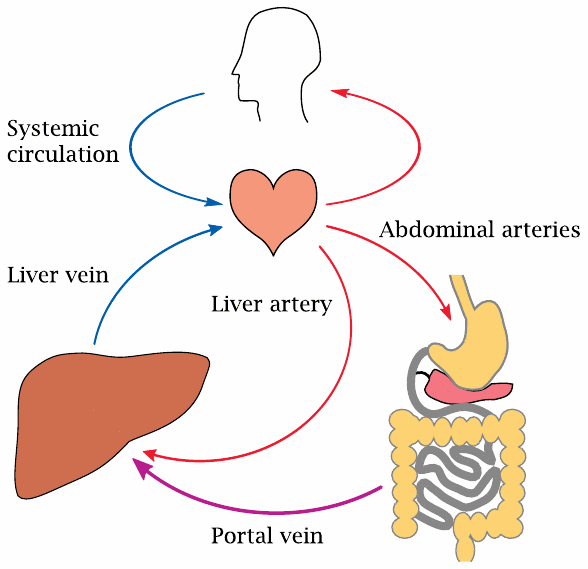
In most organs, the circulation consists of arteries, which supply oxygen-rich blood from the left heart, the capillaries, which enable gas and substrate exchange, and the veins, which drain the oxygen-depleted blood back to the right heart.
The intestinal organs—that is, the stomach, small and large intestine—as well as the pancreas and spleen also receive arterial blood from the left heart. However, their venous blood is not transported back to the heart directly; instead, it is drained into the portal vein, which then feeds it into the liver. This allows the liver to act as a checkpoint; any solutes that have been taken up in the intestines can be extracted and modified here. Following this liver passage, the blood flows back into the general circulation via the liver vein.
In addition to the already oxygen-depleted blood from the portal vein, the liver also receives a direct supply of oxygen-rich blood via the liver artery.
| 3.6.2 |
Tissue structure of the liver |
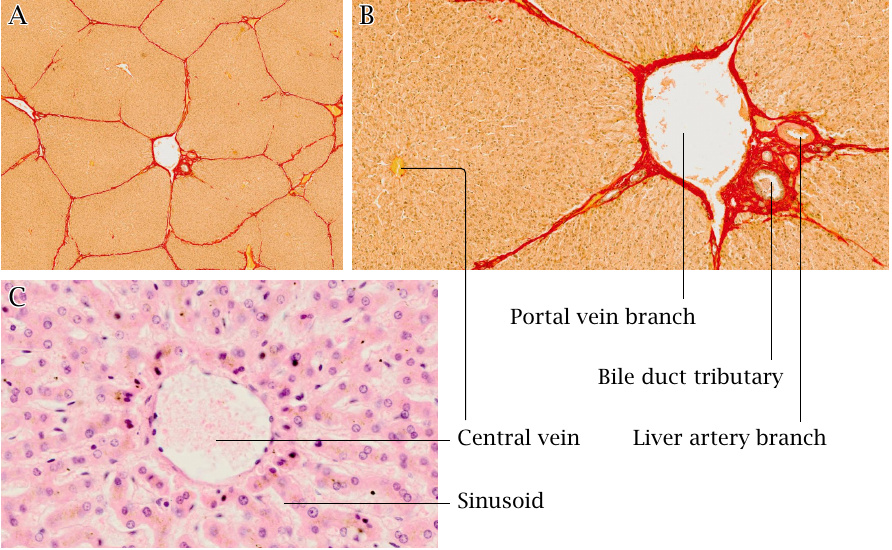
The liver tissue is organized into lobules that are a few millimeters in diameter (A).19 Each liver lobule receives blood from the terminal branches of both the portal vein and the liver artery (B). The blood leaves these branches and seeps through the hollows (sinusoids) of the liver lobule; it is collected by the central vein (C).
Next to the branches of the portal vein and liver artery, we also find tributaries of the bile duct, which drains the bile produced in the liver lobule toward the small intestine. The very thin bile conduits that originate within the lobule itself are not visible in this picture; are so thin that they can only be visualized using special histological stains or by electron microscopy.
Histological slices in A–C reproduced with permission from pathorama.ch.
| 3.6.3 |
Blood flow and bile flow in the liver lobule |
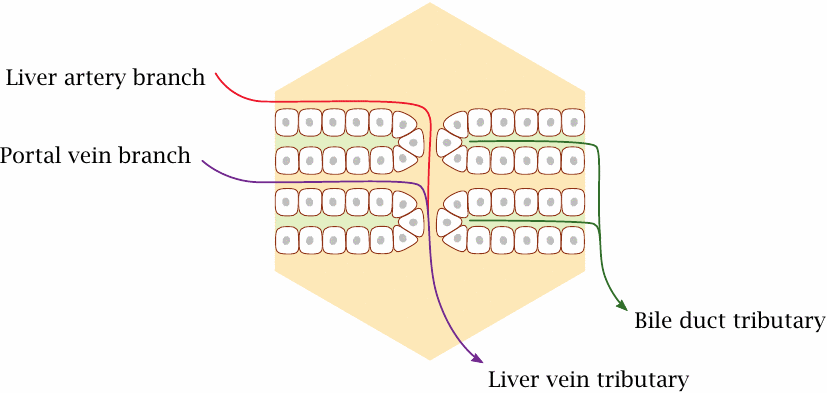
The epithelial cells in each liver lobule are arranged in parallel layers. One side of each cell faces the blood-filled sinusoid, the other faces a bile duct tributary.
The liver cells extract solutes from the blood, modify them, and export them either back into the bloodstream or directly into the bile. This process can be very efficient; with some solutes, extraction and modification are almost complete during a single pass through the liver.
| 3.6.4 |
Propranolol and the first-pass effect |
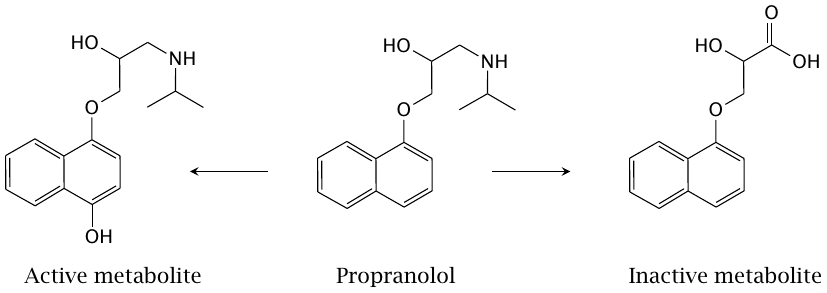
Propranolol is a β-adrenergic receptor antagonist propranolol that is used to treat some patients with cardiovascular disease. During its initial passage through the intestine and the liver, it undergoes metabolic transformation to an extent of >50%. The two metabolites shown here are formed by cytochrome P450 enzymes. The metabolite on the left (4-hydroxypropranolol) has a pharmacological activity similar to propranolol; it is an active metabolite. In contrast, naphthyloxylactate (right) is inactive.
While a first pass effect of >50% may seem high, it is not uncommon, and propranolol is indeed nevertheless given orally.
| 3.6.5 |
Outline of lung anatomy |
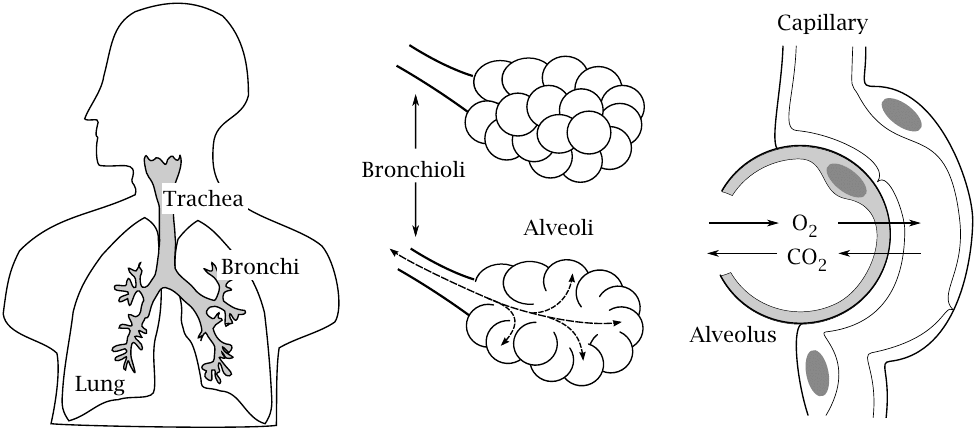
For gaseous drugs, most commonly inhalation anesthetics, inhalation is the most straightforward route of application and results in very rapid uptake. The efficiency of uptake is due to the organ structure of the lung.
The trachea branches out into bronchi and then bronchioli that open out into alveoli. The alveoli collectively have a very large active surface (~80 m2), and the distance between the air-filled space and the blood in the surrounding capillaries is only a few micrometers. This combination of large area and short distance of diffusion enables a very rapid gas exchange.
| 3.6.6 |
Major compartments of drug distribution |
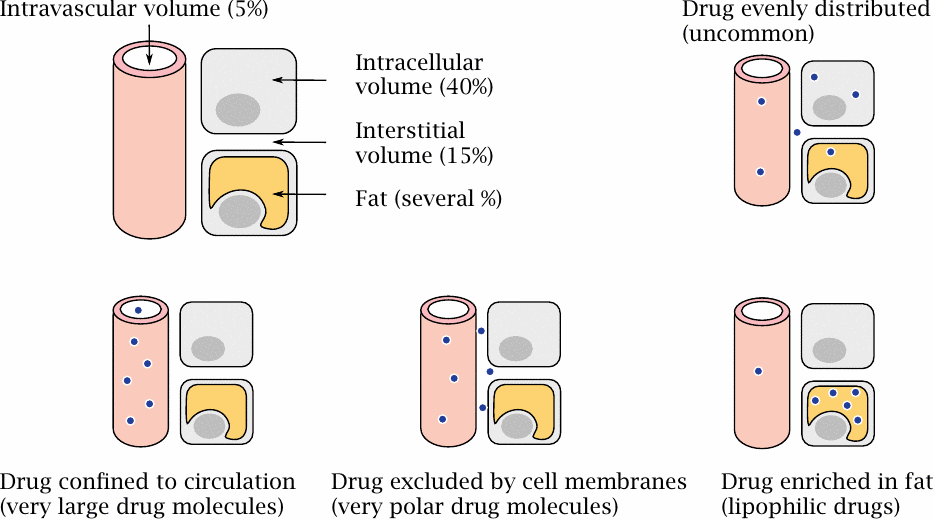
This picture summarizes the different compartments between which drugs may distribute. These spaces are not divided according to organs but rather according to tissue architecture. Drugs need to traverse different barriers on their way from one compartment to the other.
Very large drug molecules—such as proteins and polysaccharides used as plasma expanders—cannot cross the basal membranes of the capillaries to reach the interstitial space and will remain confined to the circulation; they will therefore occur in the blood plasma at high concentrations. On the other hand, drugs that are small enough to leave the capillaries and apolar enough to easily cross cell membranes will have low plasma concentrations, particularly when they are very lipophilic and accumulate inside fat cells.
One useful, easily measured parameter that characterizes the distribution of a drug is the volume of distribution (Vd). It is defined as the ratio of the drug’s number of molecules in the body (which is known from the total dosage) divided by the plasma concentration; therefore, it varies inversely with the fraction of the drug that is present in the blood plasma. The name of this parameter only reflects the fact that it has the physical dimension of a volume; it is not an actual volume. Indeed, some very lipophilic drugs have Vd values that are many times greater than the actual body volume. More often than not, the volume of distribution is normalized to the body weight and therefore stated in units of l/kg; even though this form no longer has the dimension of a volume, it is still called by the same name.
| 3.6.7 |
Hydrophobic drugs tend to bind to proteins |
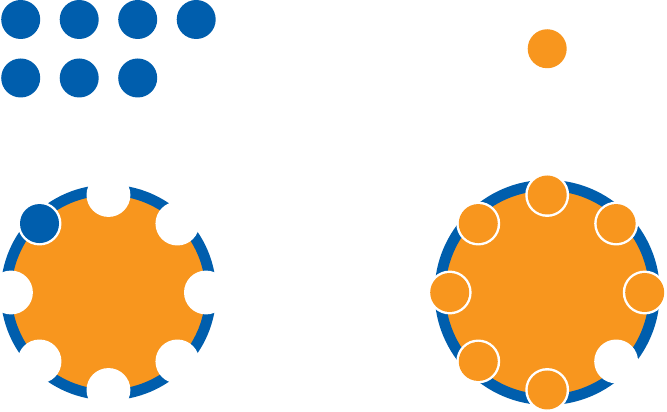
The surfaces of protein molecules are mostly hydrophilic, but they often have hydrophopic patches and crevices that may bind hydrophobic drug molecules. Protein binding can also be mediated by other molecular interactions such as hydrogen bonds and Coulomb forces.
Protein binding is particularly common in blood plasma, which has a very high protein content (~60–80 grams per liter). Two thirds of the total plasma protein is made up by a single protein, namely albumin. Each albumin molecule has eight hydrophobic binding pockets, which in regular physiology serve to transport free fatty acids. These binding pockets also accommodate hydrophobic drug molecules.
Protein binding delays the distribution from the plasma into the tissues. I also slows down filtration in the kidneys (see below), and it thus tends to delay the elimination of drug molecules.
| 3.6.8 |
Example drugs and their uptake and distribution parameters |
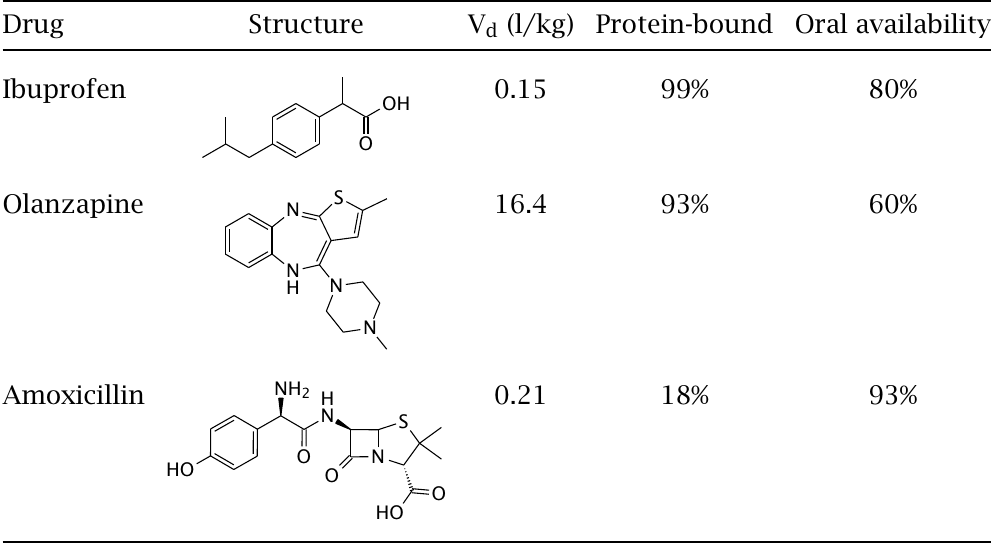
Ibuprofen and olanzapine are fairly hydrophobic, whereas amoxicillin is more hydrophilic. Nevertheless, all three drugs are readily taken up from the intestine; amoxicillin even has the best oral availability.20 Note that the oral availability is not only affected by the traversal of the intestinal mucous membrane but also by the hepatic first-pass effect.
Olanzapine has a very high Vd value, suggesting that it accumulates in some sort of sink outside of the circulation, most likely inside fat cells. On the other hand, both ibuprofen and amoxicillin have low Vd values, suggesting that they are largely confined to the extracellular compartment. In the case of ibuprofen, a fairly high blood plasma level is likely caused by strong binding to albumin.21
The ineffective penetration of the intracelluar compartment observed with amoxicillin is also observed with many other penicillins and with cephalosporins, and it limits the usefulness of these drugs with bacterial pathogens such as Listeria monocytogenes that persist intracellularly.
| 3.6.9 |
Kinetics of thiopental distribution |
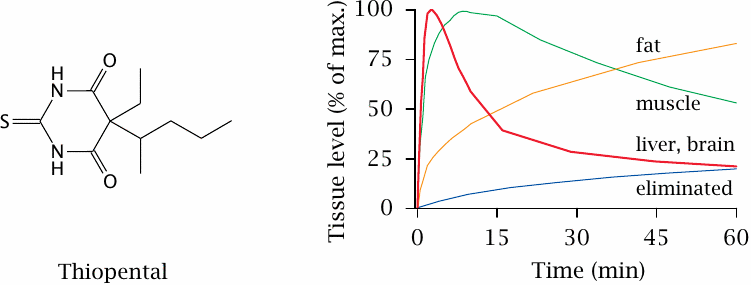
In our above treatment of drug distribution, we implicitly assumed that drugs equilibrate readily between the different compartments, and we neglected distinctions between different organs. One of these neglected distinctions between organs concerns their different relative rates of blood perfusion.
With the drug thiopental, the unequal rates of perfusion create a characteristic time course of distribution that is essential for this drug’s clinical use, namely the induction of a short-lived state of narcosis.
The plot on the right shows the time course of thiopental levels in various tissues after intravenous application. After a very short initial phase of accumulation in the lung (not shown), thiopental redistributes to the next most strongly perfused organs such as the brain and the liver. It is during this phase that the patient, for a few minutes, experiences (or rather, doesn’t experience) narcosis.
From the brain, the drug then redistributes to skeletal muscle—which, in a resting patient, is not very strongly perfused—and finally to fat tissue, which has the lowest rate of perfusion but accumulates the drug because of its lipophilicity. Ultimate elimination from the body is very slow, but this does not affect the time course of the narcotic effect. Figure prepared from original data in [16].
| 3.7 |
Kidney function and drug elimination |
The kidneys and the liver share in the work of getting rid of drugs and their metabolites. Drug elimination by the kidneys will be covered in this chapter; the role of the liver is considered in the next chapter.
| 3.7.1 |
Overview of drug elimination |
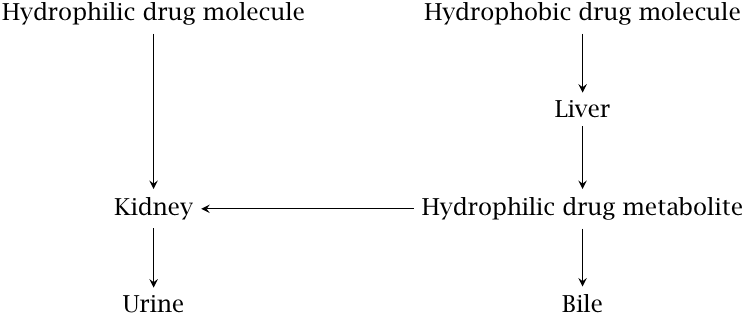
Very broadly speaking, hydrophilic drug molecules tend to be eliminated via the kidneys directly, whereas hydrophobic drugs require metabolic transformation in the liver before undergoing renal or biliary elimination. There are, however, numerous exceptions to this rule.
| 3.7.2 |
Location and perfusion of the kidneys |
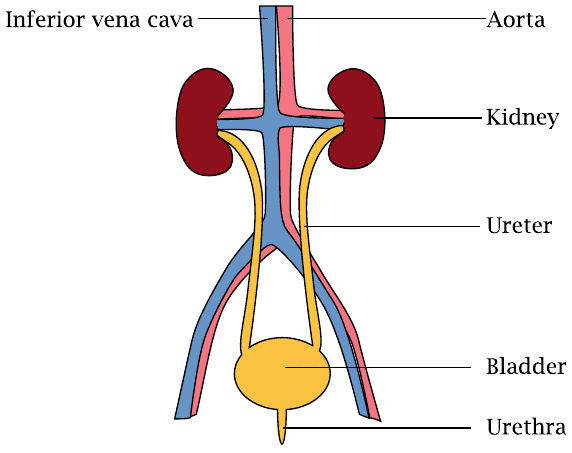
The kidneys are situated close to two major blood vessels, the aorta and the inferior vena cava. In terms of blood flow per tissue mass, the kidneys are the most strongly perfused organs in the human body. This high rate of blood flow is an important aspect of kidney function.
| 3.7.3 |
Overview of kidney function |
Urine is “distilled” from blood plasma in several stages:
- Ultrafiltration: 10-20% of the blood plasma volume flow is squeezed out; macromolecules are retained
- Solute reuptake: glucose, salts, amino acids etc. are recovered from the primary filtrate by active transport
- Water reuptake: driven by osmotic gradient
- Solute secretion: some substrates are actively secreted into the nascent urine
Blood flows through the kidneys at a rate of ~1.2 l/min, or ~1700 l/day. A bit more than half of that volume is blood plasma, and if we assume a filtration rate of 15%, we obtain a filtrate volume of approximately 150 l/day. Obviously, therefore, most of the water and of the solutes contained in the filtrate are reclaimed, and only the leftovers appear in the final urine. On the other hand, some solutes are secreted into the urine only after filtration.
Kidney physiology is fascinating, and I encourage you to read about it in a good physiology textbook. Here, I will not try to give a complete picture, but focus on the aspects that are most relevant to drug elimination.
| 3.7.4 |
The nephron |
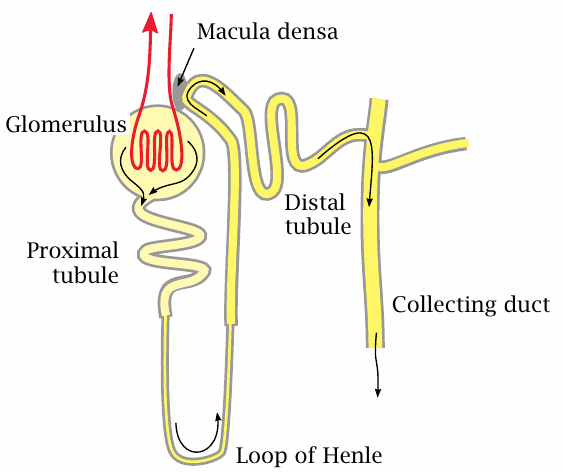
Just as the liver consists of many lobules that each comprise the whole organ function in a small microcosm, the kidney consists of many instances of a repeating functional unit, the nephron, that comprises all stages of urine production. Each of the two kidneys contains about 1.3 million nephrons, all working in parallel.
A nephron consists of a glomerulus and a tubular part that can be divided into the proximal tubule, the loop of Henle, and the distal tubule; the latter joins a collecting duct that drains several nephrons.
The primary filtrate is formed in the glomerulus. It flows into the proximal tubule, where glucose, amino acids, and most of the salt ions are reclaimed by specific active transporters; water follows by osmosis. More water is reclaimed in Henle’s loop. Along this stretch, a very high salt concentration prevails in the interstitial space of the surrounding tissue, which allows the nascent urine to become significantly more concentrated than blood plasma. Fine tuning of concentration, pH, and salt content occurs in the distal tubule.
| 3.7.5 |
Cross sections of glomeruli and tubules in kidney |
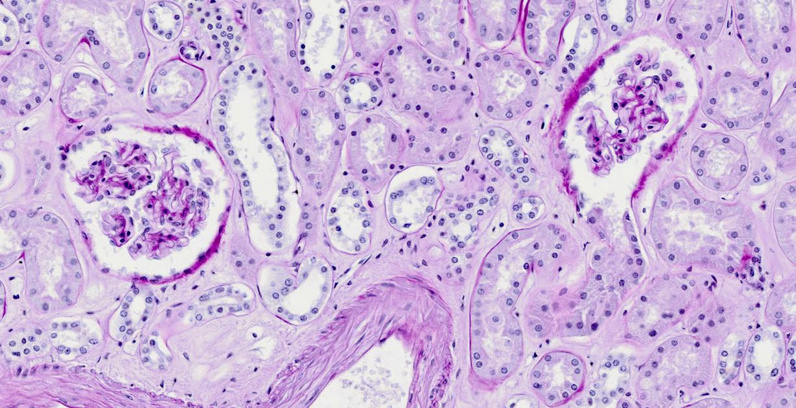
The tissue slice (from pathorama, with permission) shows two glomeruli, surrounded by cross sections of various types of tubules. The sheath containing each glomerulus is known as Bowman’s capsule. In the glomerulus on the right, the capsule has been cut away where it opens into the proximal tubule.
| 3.7.6 |
Plasma ultrafiltration in the glomerulus |
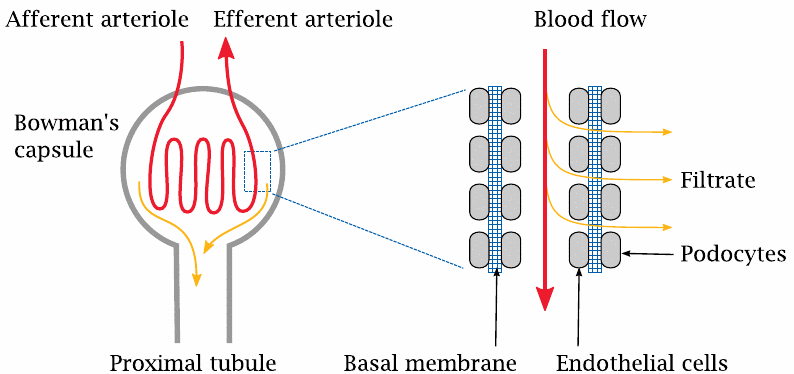
The glomerulus contains a coiled arteriole, that is, a small artery. Unlike the arterioles in the remainder of the body, the endothelium of the glomerular arterioles has gaps; these line up with gaps in a second cellular layer, formed by the so-called podocytes, that surrounds the outer side of the basal membrane of the arteriole. As with the capillaries in the general circulation, it is the basal membrane that forms the actual molecular sieve in the filtration. The molecular weight cutoff for filtration is similar, too; molecules with less than 10 kDa get across, larger ones are retained. For small molecules that are not retained in the plasma by protein binding, the concentration in the filtrate will be the same as in the plasma.
Glomerular filtration is driven by the hydrostatic pressure gradient across the wall of the arteriole. In capillaries, the hydrostatic pressure is fairly low, and since it is almost entirely balanced by the osmotic activity of the plasma proteins, very little net filtration occurs. The pressure within the glomerular arterioles is much higher, and therefore filtration proceeds apace.
| 3.7.7 |
Filtration, reuptake, and active secretion |
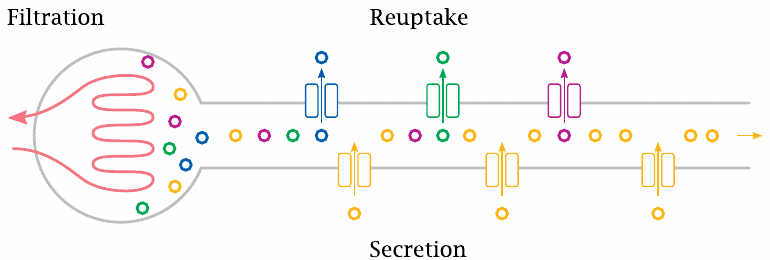
The glomerular filtrate is extensively post-processed as it passes down the tubule. Many solutes are recovered from the filtrate, whereas some other solutes are secreted into the nascent urine by specific active transport.
Drugs can be substrates at all three stages; that is, they can be subject to filtration, reuptake, and secretion.
| 3.7.8 |
A drug’s rate of urinary excretion depends on its membrane permeability |
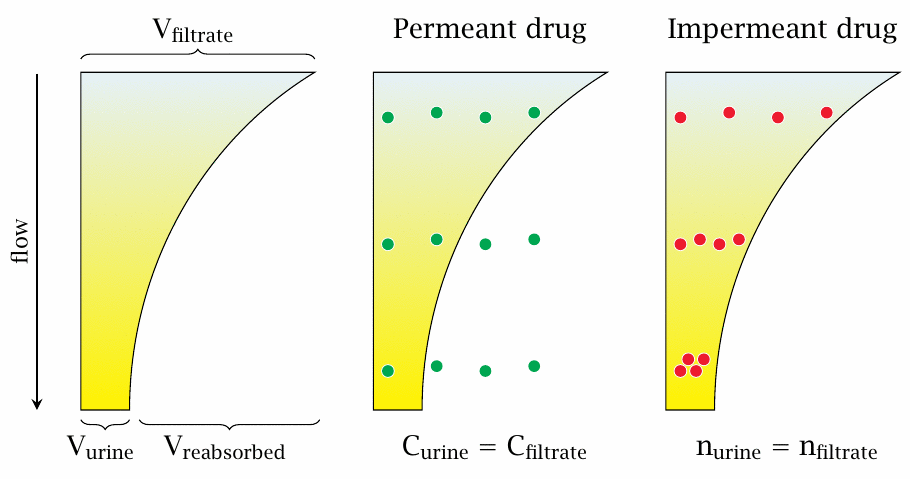
As we had seen before, most of the water is recovered from the primary filtrate as it passes down the tubules. As the volume of the filtrate diminishes, the concentration of a drug contained in it increases, creating a gradient between the tubule and the surrounding interstitial space. Whether or not (or the extent to which) the drug will travel downhill this concentration gradient, across the tubular epithelium, depends on its membrane permeability.
The slide depicts the two limiting cases. A fully membrane-permeant drug may freely equilibrate between the tubule and the surrounding interstitial fluid. Its concentration in the urine will therefore be the same as in the interstitial fluid, and the rate of its urinary excretion will be determined simply by the product of this concentration and the urine volume flow.22
On the other hand, a completely impermeant drug will be quantitatively retained in the nascent urine. Therefore, the absolute number of drug molecules remains the same between the primary filtrate and the final urine, and the urine concentration exceeds the filtrate concentration in inverse proportion to the volume fraction of the filtrate that makes it to the final urine. Furthermore, the rate of elimination will depend only on the rate of glomerular filtration, but not on the urine volume flow rate.
We had previously encountered the principle of non-ionic solute diffusion across lipid bilayers (see slide 3.4.7). We can use this principle to advantage in order to accelerate the renal elimination of certain drugs. Apart from xenobiotics and metabolic end products, the kidneys also eliminate excess acid or base. Through the application of (buffered) acid or base, we can therefore induce the kidneys to lower or raise the urine pH. This can for example be used in phenobarbital poisoning. This drug (see structure on slide 4.1.4) is a weak acid. To accelerate its elimination, we can raise the urine pH using sodium bicarbonate infusions. This will cause phenobarbital in the urine to be deprotonated, which will inhibit its escape by non-ionic diffusion and thereby promote its excretion [17].23
| 3.7.9 |
Inulin, a model compound that is quantitatively filtrated and retained in the urine |
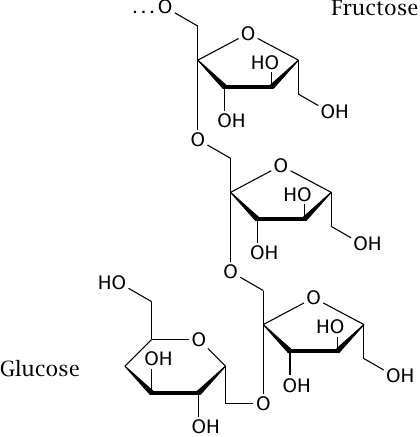
Inulin is a polysaccharide that is small enough to undergo quantitative filtration in the glomeruli. It is also polar enough to avoid protein binding and reuptake by diffusion across bilayers; therefore, it represents the second limiting case presented in slide 3.7.8). Based on these properties, inulin can be used to non-invasively measure the volume flow rate of glomerular filtration.
| 3.7.10 |
The volume flow of glomerular filtration can be measured from the renal clearance of inulin |

This slide shows what parameters we require in order to measure the glomerular filtration rate (GFR; dV filtrate/dt). The flow rate of the urine can be obtained simply by collecting the urine for 24 hours. The concentrations of inulin in the urine and in the filtrate and the blood plasma are obtained with chemical methods. This analysis assumes steady state for the inulin plasma concentration, which can be established by continuous infusion. Since inulin is quantitatively filtrated, we can equate the experimentally determined plasma concentration with the concentration in the filtrate.
The last term in the last equation—urine volume flow times urine concentration over plasma concentration—is referred to as the renal clearance of a compound. The inulin clearance equals the GFR because inulin is quantitatively filtrated but not subject to tubular secretion or reuptake. Most other compounds don’t meet all of these criteria, and thus their renal clearances do not equal the GFR. Nevertheless, the GFR is a major determinant in the elimination of many drugs, and it therefore is of considerable interest in pharmacokinetics and pharmacotherapy.
In clinical practice, the GFR is measured not with inulin but with the endogenous metabolite creatinine. This metabolite is formed at a fairly constant rate in skeletal muscle, obviating the need to administer an infusion. While the behavior of creatinine is not as “ideal” as that of inulin, the creatinine clearance provides an estimate for the GFR that is good enough for the practical need of adjusting dosage regimens for drugs that undergo renal elimination to the kidney function of individual patients.
| 3.7.11 |
Tubular secretion of p-aminohippurate |
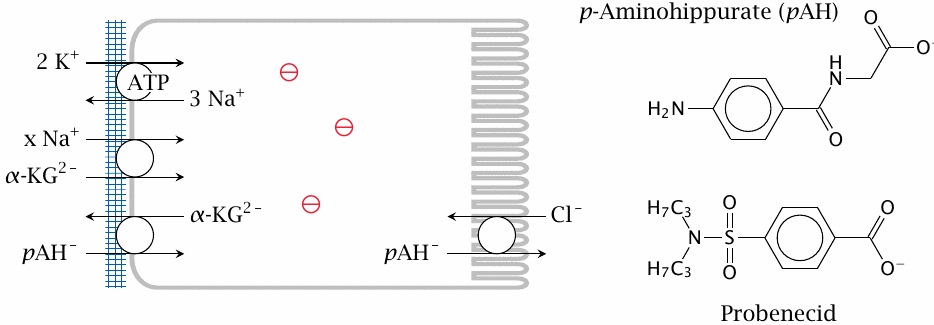
A model compound for studying the tubular secretion of drugs is p-aminohippurate (p-AH). Import of p-aminohippurate from the interstitial space, across the basolateral membrane of the cells of the tubular epithelium, is driven by exchange for α-ketoglutarate (α-KG). The required α-ketoglutarate gradient is maintained by Na+-symport, and the Na+ gradient in turn is maintained by Na+/K+-ATPase. The excretion of p-aminohippurate across the apical membrane into the tubular lumen is driven by antiport with Cl− or HCO3−. This step can be inhibited by the drug probenecid.24
The free energy of solute transport across the basolateral membrane is affected not only by concentration gradients but also by the negative-inside membrane potential, which makes the exchange of the singly negative p-aminohippurate for the doubly negative α-ketoglutarate more exergonic.
| 3.7.12 |
Tubular secretion of p-aminohippurate is almost quantitative |
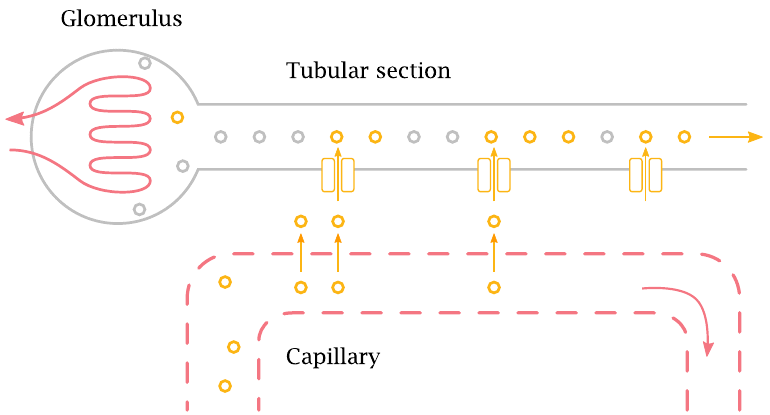
In each nephron, blood vessels run alongside the tubular segments to facilitate the efficient transfer of solutes from the blood to the nascent urine. The solutes permeate out of the capillaries and are then picked up by transport proteins that eject them into the tubules.
In the case of p-aminohippurate, the transport rate is so high that ≥ 90% of the compound is extracted from the blood and transferred to the urine in a single passage of the blood through the kidney.
| 3.7.13 |
The p-aminohippurate clearance measures the renal plasma flow |
| \(\frac{\text{dn}_{\,p\text{-AH, plasma}}}{\text{dt}}\) | \(\approx\) | \(\frac{\text{dn}_{\,p\text{-AH, urine}}}{\text{dt}}\) | |
| np-AH | = | \(\text{[p\text{-AH}]} \times \text{V}\) | |
| \(\frac{\text{dV}_{\,\text{plasma}}}{\text{dt}} \times [p\text{-AH}]_{\text{plasma}}\) | \(\approx\) | \(\frac{\text{dV}_{\,\text{urine}}}{\text{dt}} \times [p\text{-AH}]_{\text{urine}}\) | |
| \(\frac{\text{dV}_{\,\text{plasma}}}{\text{dt}}\) | \(\approx\) | \(\frac{\text{dV}_{\,\text{urine}}}{\text{dt}}\times \frac{[p\text{-AH}]_{\text{urine}}}{[p\text{-AH}]_{\text{plasma}}}\) |
Since p-aminohippurate is almost quantitatively extracted from the plasma, we can use its clearance to measure the renal plasma flow. The first equation simply states that, over time and at steady state, the number of p-aminohippurate molecules appearing in the urine is approximately equal to the number of molecules carried to the kidneys with the plasma flow. The second equation expands the number of solute molecules into the product of concentration and volume, and the third equation substitutes both terms in the first equation according to the second. The fourth equation is obtained by rearranging the third; it tells us that the renal plasma flow is approximated by the p-aminohippurate clearance.
The practical side of determining the plasma flow from the p-aminohippurate clearance is analogous to the use of inulin clearance to measure the filtrate flow: Steady state is established using a continuous infusion of p-aminohippurate, urine is collected, and plasma and urine concentrations are measured.
| 3.8 |
Non-equilibrium kinetics of drug elimination |
| n | = | \(_{\text{plasma}}\times V_{\text{d}}\times \text{body weight}\) | |
| \(\mathrm{\frac{dn}{dt}}\) | = | \(-k\times[\text{D}]_{\text{plasma}}\) | |
| \(\frac{[\text{D}]_{\text{plasma,t}}}{[\text{D}]_{\text{plasma,0}}}\) | = | \(e^{ -\frac{k} {V_{\text{d}} \times \text{body weight}} t }\) | |
| 0.5 | = | \(e^{ -\frac{k} {V_{\text{d}} \times \text{body weight}} t_{\nicefrac{1}{2}} }\) | |
| \(t_{\nicefrac{1}{2}}\) | = | \(\ln2\times\frac{V_{\text{d}}\times\text{body weight}}{k}\) |
The elimination rates of inulin and p-aminohippurate were examined at steady state; however, a practically more important case is the elimination of a drug that is applied only once or intermittently. In the simplest case, elimination then follows first-order kinetics. This case is also very common in practice.
The first equation simply states the number of drug molecules in the body; Vd is the volume of distribution in units of liters per kilogram of body weight. The second equation postulates first order kinetics, that is, it states that the rate of elimination is proportional to the plasma concentration. The kinetic constant k is the big unknown here: factors like protein binding, which prevents filtration, tubular secretion, and membrane permeability, which promotes nonspecific reuptake, will all be comprised in this rate constant.
The third equation is obtained by integration of the second and substitution using the first, and the fourth one by taking the logarithm; \(t_{\nicefrac{1}{2}}\) is the half-life of elimination. While this does not get rid of k, the big unknown, it is interesting to note that the half-life of elimination increases with the volume of distribution. This makes sense: filtration or tubular secretion can only operate on the fraction of the drug that currently resides in the circulation. A high Vd implies that this circulating fraction is small, which slows down elimination.
Note that our equations make no assumptions regarding the mechanism of the elimination—it might be filtration, secretion, even metabolic transformation in the liver, or some combination of the above. Indeed, the concepts of clearance and half-life are useful with any drug, regardless of its route of excretion.
| 3.8.1 |
Repeated drug application can result in accumulation |
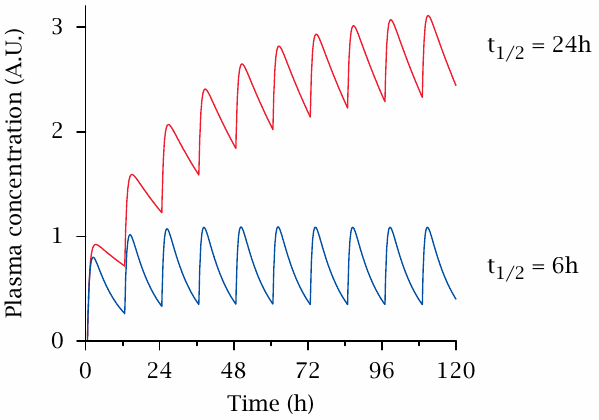
In practical pharmacotherapy, we typically want to keep the concentration of a drug within its therapeutic range throughout, which means that we have to repeat its application before the last dosage has been completely eliminated. Such repeated application will cause accumulation, the extent of which will depend on the dosage interval and the drug’s half-life of elimination. In the slide, this is illustrated for two hypothetical drugs, which have half-lives of 6 hours and of 24 hours, respectively. With both drugs, application was assumed to occur in intervals of 12 hours, and absorption with a half life of 0.5 hours. It is clear that, all else being equal, a longer half-life of elimination gives rise to a greater extent of accumulation.
Accumulation through repeated application needs to be taken into account with drugs that have a small therapeutic range. One way to deal with this is to use a larger initial loading dose that brings the concentration to the therapeutic level straight away, followed by smaller repeating doses that are calculated to maintain the level established by the initial one. While this approach is logical in principle, I have rarely seen it used in practice.