| 10 |
Intermediate metabolism, diabetes, and atherosclerosis |
| 10.1 |
Overview |
- Genetic enzyme defects
- rare; comparatively little effort spent on targeted drug development; only a few defects can be treated with drugs
- Gout
- more common; multiple causes, similar manifestation and treatment
- Diabetes mellitus
- very common; treatment with insulin injections (types 1 and 2) and oral antidiabetics (type 2)
- Atherosclerosis
- exceedingly common; drug therapy targets underlying metabolic conditions, other risk factors, and consequences of advanced disease
Intermediate metabolism involves numerous enzymes, and inheritable defects for many of them may cause disease. Only in a few cases can these genetic diseases be treated with drugs, and therefore, it does not make sense here to treat the entirety of metabolism in a systematic manner. Instead, we will look at a few selected defects for which some form of pharmacological treatment is available.
Diabetes is caused by endocrine dysregulation rather than primarily by enzyme deficiencies, so it could as well have been covered in chapter 7. It was included here because its manifestations are mostly metabolic. Also somewhat arbitrarily, atherosclerosis is covered here because of its important connections to cholesterol metabolism.
| 10.2 |
Enzyme defects in amino acid metabolism |
The two hereditary enzyme defects that we will discuss here, phenylketonuria and tyrosinemia, both affect the same pathway, namely the degradation of phenylalanine and tyrosine. Their clinical manifestations are quite different, however, and so are their treatments.
| 10.2.1 |
Degradation of phenylalanine and tyrosine |
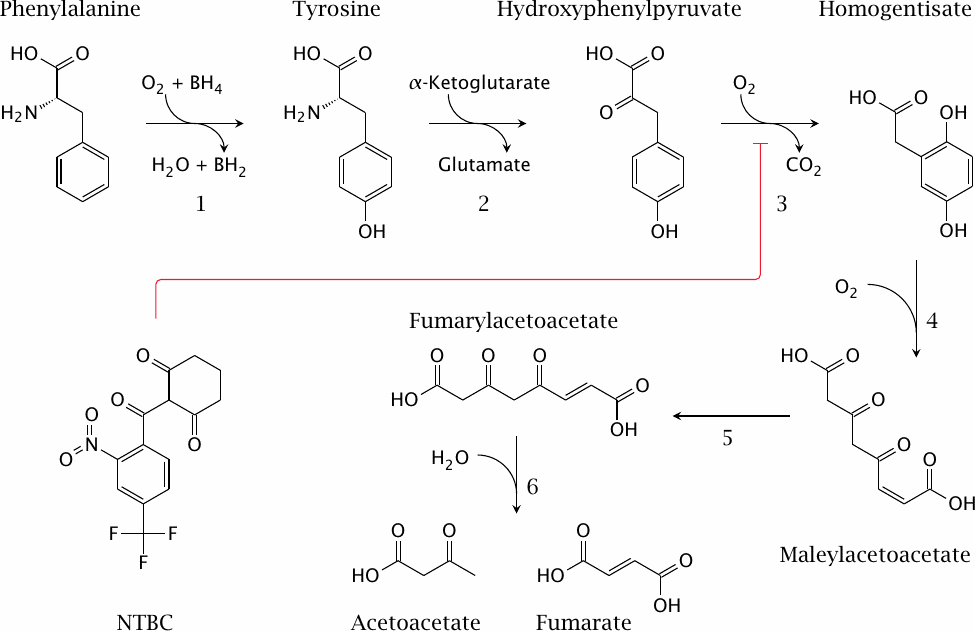
This slide shows the degradation pathway for phenylalanine and of tyrosine, and the structure of NTBC (2-[2-nitro-4-(trifluoromethyl)benzoyl]cyclohexane-1,3-dione), an inhibitor of p-hydroxyphenylpyruvate dioxygenase (3) that is used in tyrosinemia type I. The enzyme defect in phenylketonuria concerns phenylalanine hydrolase (1), and the one in tyrosinemia affects fumarylacetoacetate hydrolase (6).
| 10.2.2 |
Phenylketonuria (PKU) |
- Homozygous defect of phenylalanine hydroxylase
- Frequency: 1 newborn among 10,000 in Caucasians; lower frequency in other races
- Possible heterozygote advantage: reduced fetal susceptibility to ochratoxin A
- Symptomatic excess of phenylalanine manifest only after birth; intrauterine development normal
- Cognitive and neurological deficits, probably due to cerebral serotonin deficit
- Treated with phenylalanine-restricted diet
- Some cases due to reduced affinity of enzyme for cofactor tetrahydrobiopterin (BH4), can be treated with high dosages of BH4
As with most genetic enzyme defects, the clinical disease is manifest only in homozygous individuals. Dietary phenylalanine that is not used for protein synthesis accumulates and causes toxicity. It appears that the excess phenylalanine crowds out tryptophan at the l-aromatic amino acid transporter in brain capillaries, which is the same transporter that also transports tyrosine and l-DOPA to the brain (see slide 3.5.5). Since tryptophan is the precursor of serotonin (see slide 6.12.1), this results in a lack of cerebral serotonin [77], which is believed to cause the observed cerebral deficits.
In addition to phenylalanine itself, some aberrant metabolites derived from it also occur at increased levels, and the appearance of ketone derivatives such as phenylpyruvic acid in the urine has given the disease its name. These metabolites have no proven connection to the pathogenesis of the disease.
| 10.2.3 |
Ochratoxin A inhibits phenylalanyl-tRNA synthetase |
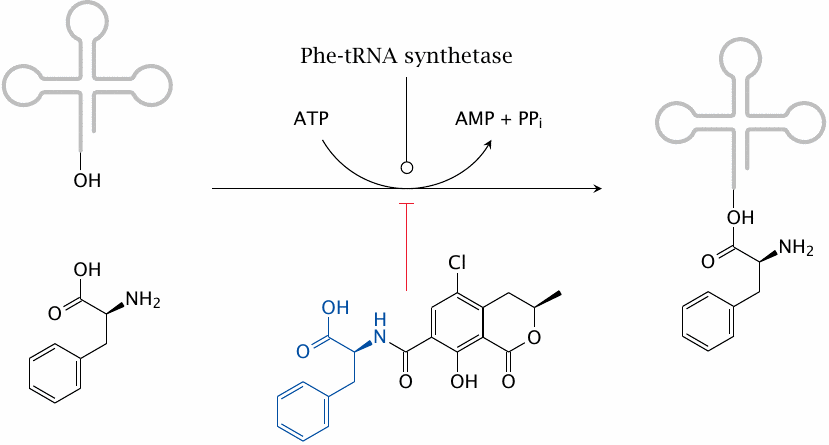
The variation of the gene frequency for PKU between races and geographical areas suggests that, in some parts of the world, environmental conditions may confer a selective advantage of the heterozygous state; accepted examples of such heterozygote advantage are sickle cell anemia and glucose-6-phosphate dehydrogenase deficiency, which endow heterozygotes with greater resistance to malaria. It has been proposed that the heterozygote advantage in PKU consists in protection from the fungal toxin ochratoxin A, which is produced by some Aspergillus molds that cause food to rot [78].
Ochratoxin A competitively inhibits the coupling of phenylalanine to its cognate tRNA and thereby disrupts protein synthesis. It is more toxic to fetuses than to adults, most likely because fetuses are short of enzymes required for detoxifying xenobiotics. Mothers who are heterozygous for PKU will have a somewhat higher level of phenylalanine, which will be shared with the fetus via the placenta. This will counter the inhibition of tRNA aminoacylation in the fetus and thereby afford it a degree of protection.
One of the places with the highest abundance of PKU is Ireland. This country is also known for its repeated historic episodes of famine. Starving people will more likely eat rotten food rather than discard it. Indeed, reference [78] reports that lowered rates of abortion were found in Irish women who were heterozygous for PKU. I have not tried to ascertain whether these quoted statistics applied to periods of actual famine.
| 10.2.4 |
Tyrosinemia |
- Homozygous defect of fumarylacetoacetate hydrolase
- Prevalence: 1 in 100,000 people worldwide; 1 in 1,850 in the Saguenay region (Quebec)
- Fumarylacetoacetate and preceding metabolites back up
- Fumaryl- and maleylacetoacetate react with glutathione and other cellular nucleophiles, causing liver toxicity, cirrhosis, carcinoma
- The drug NTBC inhibits p-hydroxyphenylpyruvate dioxygenase, intercepting the degradative pathway upstream of the toxic metabolites
Tyrosinemia is comparatively common in Quebec. In this case, there seems to be no heterozygote advantage; instead, the high incidence is due to the so-called “founder effect”, that is, the common descent of the afflicted population from a small group of founding settlers that happened to contain one or several carriers of the gene. (Refer to slide 10.2.1 for the relevant pathway and enzyme reactions.)
| 10.2.5 |
The urea cycle and related pathways |
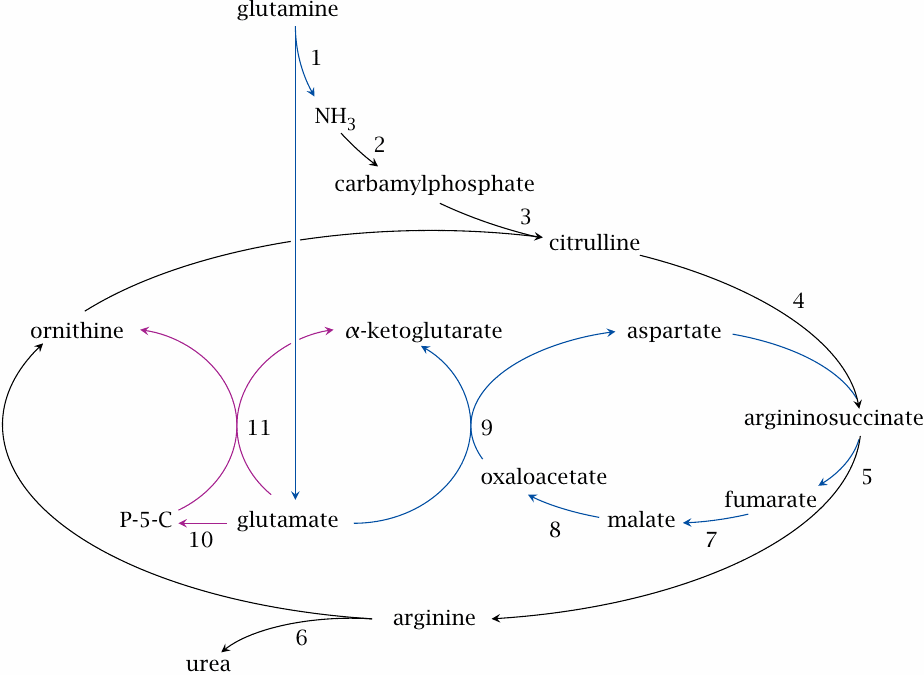
The urea cycle runs in the liver and disposes of ammonia that accrues in the degradation of amino acids. It incorporates two molecules of ammonia into one molecule of urea, which is excreted in the urine.
While amino acid degradation occurs to a large part in the liver, other organs also participate; for example, the branched-chain amino acids leucine, isoleucine and valine are mostly degraded in skeletal muscle. Surplus ammonia is transported from these organs to the liver in the form of alanine or glutamine.
In the slide, reactions 2–6, shown in black, form the urea cycle. Reactions 1 and 7–9, shown in blue, supply the urea cycle with nitrogen from glutamine. Reaction 10 yields Δ1-pyrroline-5-carboxylate (P-5-C), and together with reaction 11 replenishes ornithine.
Enzyme defects occur in various reactions of the urea cycle; all of them will cause the accumulation of surplus ammonia. The enzyme defects are treated with protein-restricted diet, alternate pathway therapy, and supplementation of arginine or citrulline.
| 10.2.6 |
Alternate pathway therapy in urea cycle enzyme defects |
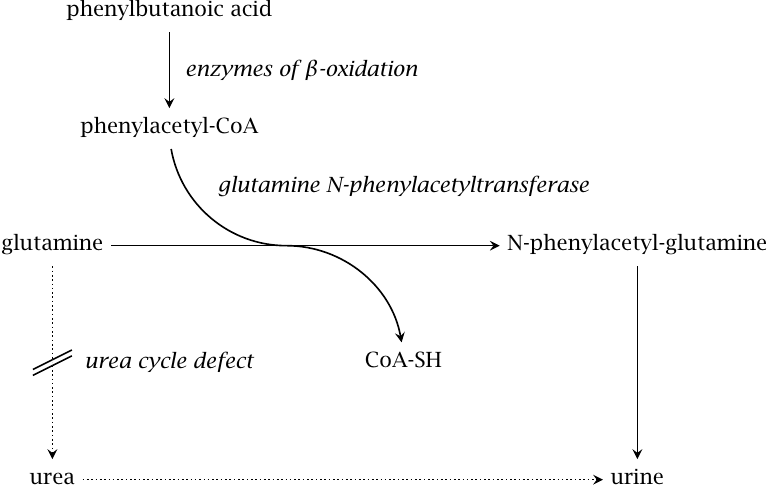
Alternate pathway takes advantage of the amino acid conjugation reactions that occur as part of phase II drug metabolism (see slide 4.4.9). In this application, the usual physiological role of amino acid conjugation is turned on its head: While in normal metabolism amino acids are expended in order to inactivate and eliminate unwanted organic acids, we here supply innocuous organic acids in order to induce their conjugation with amino acids. The elimination of these conjugates then becomes an alternate means for the disposal of surplus nitrogen. Brilliant!
The substrate acid covered in this scheme is phenylbutanoate, which undergoes β-oxidation before conjugation to glutamine and excretion in the urine. Another useful substrate acid is benzoate, which undergoes conjugation with glycine and further drains the liver’s pool of nitrogen.
Alternate pathway therapy is combined with the application of arginine or citrulline. If the urea cycle is functional, arginine can be diverted to protein synthesis as required, and so is not an essential amino acid; however, disruption of the urea cycle may cause a shortage of arginine. This may induce protein catabolism, thereby exacerbating the symptoms. If the enzyme defect is not between citrulline and arginine, it is possible to supply citrulline instead, which has the advantage of picking up another surplus nitrogen en route to arginine.
| 10.3 |
Metabolic diseases related to purine nucleotide metabolism |
In this section, we will consider adenosine deaminase deficiency and gout. Adenosine deaminase deficiency is a hereditary enzyme defect and, as such, a rare condition. In contrast, gout can arise from multiple causes and thus is much more common.
| 10.3.1 |
Overview of purine degradation |
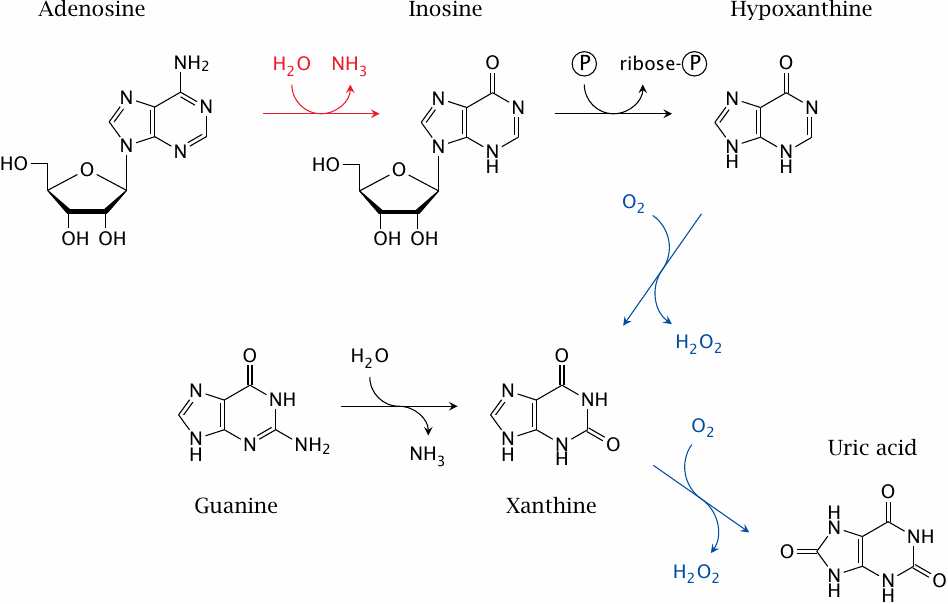
This slide shows some of the reactions involved in purine nucleotide degradation. The reaction catalyzed by adenosine deaminase is shown in red; those catalyzed by xanthine oxidase, which is a drug target in gout, are shown in blue. Uric acid is the final degradation product that is excreted in the urine.
Note that adenosine deaminase degrades both adenosine (shown here) and deoxyadenosine. Accumulation of deoxyadenosine is crucial for pathogenesis in this disease (see next slide).
| 10.3.2 |
Adenosine deaminase deficiency causes dysregulation of DNA synthesis |
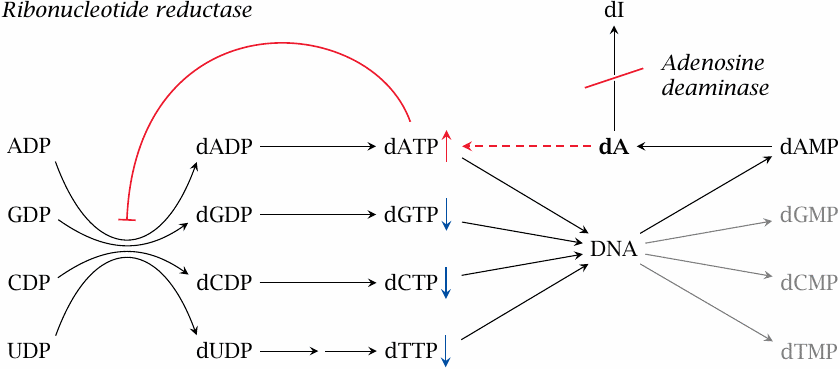
Adenosine deaminase (ADA) degrades not only adenosine but also deoxyadenosine (dA), which it converts to deoxyinosine (dI). If ADA is lacking, dA accumulates and is converted back to dATP by salvage pathway nucleoside and nucleotide kinases. Excess dATP inhibits ribonucleotide reductase and thereby throttles the supply of deoxyribonucloside triphosphates other than dATP. This interferes with DNA synthesis and promotes apoptosis.
The apoptotic stimulus created by ADA deficiency has limited strength and, in most cell types, falls short of triggering manifest apoptosis. However, it does induce apoptosis in lymphocytes, which are very susceptible to apoptotic stimuli in general. Apoptosis depletes both T- and B-lymphocytes, which gives rise to severe combined immunodeficiency (SCID).70
| 10.3.3 |
Therapy of adenosine deaminase deficiency |
- Bone marrow transplant
- Ex vivo gene therapy
- Enzyme replacement therapy—PEGylated bovine ADA
- Experimental in vitro approach: Inhibition of salvage kinases
The current standard treatment of ADA is bone marrow transplant. If the bone marrow of the patient is replaced with that of a healthy donor, the lymphocytes derived from the new bone marrow will express an intact ADA gene, and therefore be able to protect themselves from dATP accumulation and apoptosis. Gene therapy is based on the same principle, but uses replacement of the defective gene within the patients’ own bone marrow stem cells. It is transitioning from experimental to routine treatment [79].
Enzyme replacement therapy with ADA enzyme isolated from cattle is effective but inferior to bone marrow transplant. It is used mostly to bridge the time interval between diagnosis and the identification of a suitable bone marrow donor. Modification of the enzyme with polyethyleneglycol (PEG) serves to reduce its immunogenicity and extend its lifetime in the circulation.
A plausible pharmacological approach to prevent the pathology caused by deoxyadenosine accumulation would be to inhibit the salvage kinases that convert deoxyadenosine to dATP, the actual inhibitor of ribonucleotide reductase. While this approach has been demonstrated in vitro[80], I have not seen any reports describing its further development in vivo.71
| 10.3.4 |
Gout |
- Genetic or dietary factors promote increased production or retention of uric acid
- Uric acid has low solubility, and excess levels form crystalline deposits, preferentially in joints and soft tissue
- Urate crystals promote inflammation and lead to arthritis that is painful and destructive
Gout results from excess levels of uric acid, the end product of adenine and guanine degradation (see slide 10.3.1). The disease is not restricted to patients with mutations in a specific gene and can arise from different causes; it thus is far more common than any of the enzyme defects covered so far. Nevertheless, the clinical symptoms remain the same and are addressed by similar therapeutic measures.
| 10.3.5 |
Transport of uric acid in the kidneys |
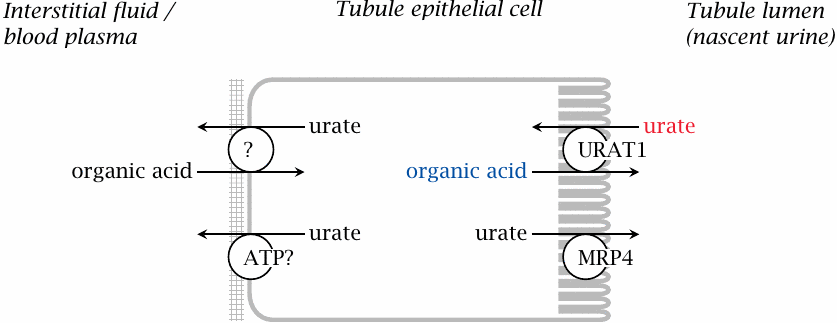
Urate is subject to glomerular filtration as well as to tubular reuptake and secretion. The key transporter in tubular reuptake is URAT1, which exchanges uric acid for other organic acids. Metabolic conditions that increase the supply of exchange substrates for URAT1 promote gout; conversely, inhibitors of URAT1 can be used to treat the disease.
| 10.3.6 |
Dietary factors that promote gout |
- Overly purine-rich food
- Drugs that contain purines: dideoxyadenosine
- Alcoholic beverages—but not all kinds: beer yes, wine no
- Anorexia nervosa (!)
- Drugs that interfere with uric acid excretion: pyrazinamide, salicylic acid
- Excessive fructose or sucrose
It seems that dietary purines are mostly not utilized via salvage pathways but instead are converted to uric acid and excreted [81]. Therefore, a diet that is overly rich in purines—typically, due to a high content of meat—offers a straightforward way to contract gout. Drugs can be a source of excess purine, too, such as for example 2,3-dideoxyadenosine (didanosine), an inhibitor of retroviral DNA synthesis that is used in HIV patients (see slide 11.10.5). The drug undergoes degradation like other purine nucleotides and may occasionally trigger gout [82].
There are several conceivable connections between alcohol and gout, of which no single one has unequivocally been shown to be the dominant one. Degradation of alcohol via alcohol dehydrogenase and then aldehyde dehydrogenase produces NADH, which shifts the equilibrium of the lactate dehydrogenase reaction from pyruvate to lactate. Lactate may then serve as an exchange substrate at URAT1 and thereby inhibit renal elimination of urate (see slide 10.3.5). However, a recent statistical study found an association of beer but not wine consumption with gout [83]. Beer is higher in calories and in purines than wine, suggesting that alcohol itself, at moderate levels of consumption, is not an important determinant.
Considering that gout is often brought on by an overly rich diet, it may be surprising that anorexia nervosa, an eating disorder in which patients eat only the bare minimum required to ward off death, and sometimes less, may also lead to gout [84]. This is probably due to the formation of ketone bodies, which as organic acids may also increase tubular reuptake of uric acid.
| 10.3.7 |
Drugs that may promote gout by promoting tubular reuptake of urate |
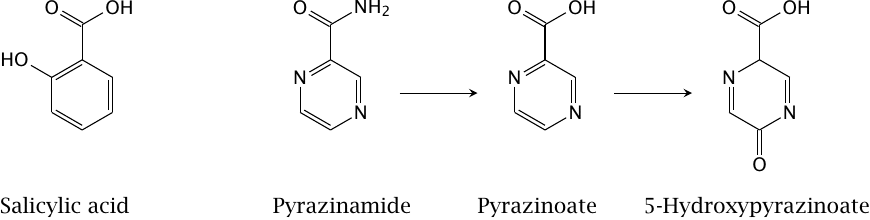
Salicylic acid as well as pyrazinoic acid and 5-hydroxypyrazinoic acid, which are formed in the metabolism of the tuberculostatic drug pyrazinamide, also act as exchange substrates for uric acid at URAT1 and thereby reduce its renal elimination. Pyrazinamide may be used in dosages of grams per day and continuously for several months, and the correspondingly large accruing amount of metabolites may result in gout.
| 10.3.8 |
High dietary fructose promotes gout |
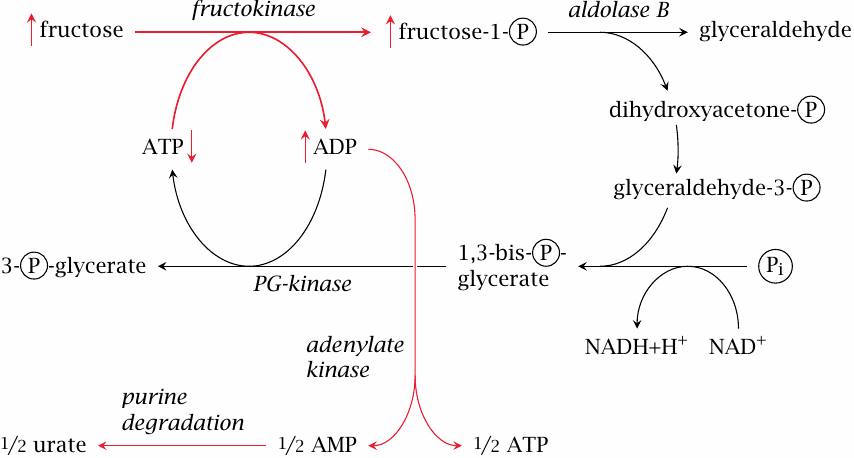
Fructose has been linked to increased uric acid production both experimentally [85,86] and statistically [87]. The mechanism is as follows: Fructokinase produces fructose-1-phosphate more rapidly than it can be turned over by aldolase B. Accumulating fructose-1-phosphate ties up phosphate, which is then no longer available for the regeneration of ATP. ADP rises, and adenylate kinase causes AMP to rise in turn; the latter enters degradation to uric acid.
In keeping with this mechanism, a fructose challenge causes a greater increase of urate synthesis in heterozygous carriers of a deficient aldolase B gene than in individuals with two intact gene copies. In the homozygous state, aldolase B deficiency causes hereditary fructose intolerance, which is also due to phosphate becoming tied up in fructose-1-phosphate, but to a much greater extent and with more immediate and potentially disastrous consequences for the liver cell.
| 10.3.9 |
Drugs used in the treatment of gout |
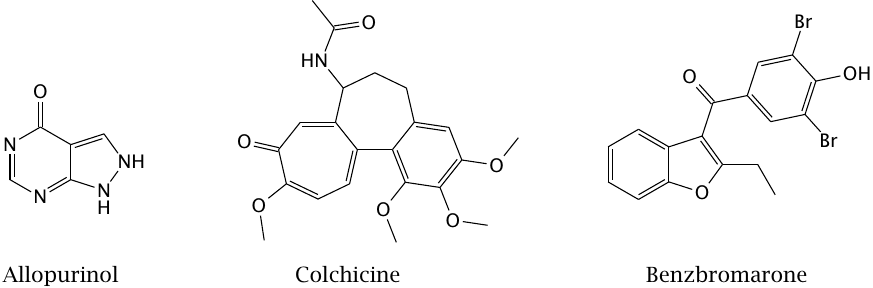
Allopurinol inhibits xanthine oxidase and thereby the formation of uric acid. Colchicine is an alkaloid that inhibits actin polymerization and has anti-inflammatory effects; it is used for symptomatic treatment in acute attacks of gout. Benzbromarone is an uricosuric drug, that is, it inhibits the tubular reuptake of uric acid by URAT1 and so increases its renal elimination.
| 10.3.10 |
Complementary effects of allopurinol and “rasburicase” |
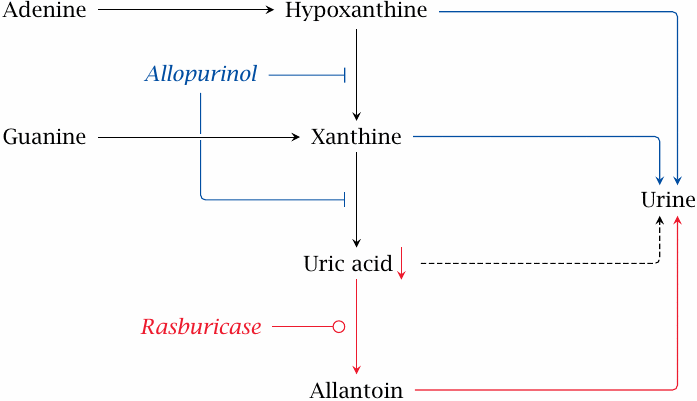
In purine degradation, adenine and guanine are converted by deamination to hypoxanthine and xanthine, respectively. Xanthine oxidase converts hypoxanthine to xanthine, as well as xanthine to uric acid. Uric acid is the final product of purine degradation and is excreted.
Uric acid has limited solubility, and increased levels of uric acid lead to its precipitation or crystallization in joints or in the kidneys. The latter occurs most acutely and dramatically in tumor lysis syndrome during chemotherapy of malignant lymphoma or leukemia.72 Tumor lysis syndrome is itself an acutely life-threatening condition.
In both gout and tumor lysis syndrome, inhibition of xanthine oxidase prevents the final oxidation steps, leading to the excretion of hypoxanthine and xanthine of uric acid. These are somewhat more soluble than uric acid and therefore less prone to precipitation prior to excretion. However, xanthine in excessive amounts can again give rise to calamitous precipitate formation.
A way out of this dilemma is the application of urate oxidase. This enzyme, which occurs in animals other than primates and in many other organisms, converts urate to allantoin, which is considerably more soluble and forms the end product of purine degradation in these organisms. “Rasburicase” is a recombinant urate oxidase preparation used to cope with excess urate in chemotherapy patients.
| 10.4 |
Lysosomal storage diseases |
- Acidic hydrolases in lysosomes break down many cellular macromolecules, including lipids and mucopolysaccharides
- Enzyme defects cause accumulation of undegraded substrates, often in liver, spleen, and bone marrow, leading to organ enlargement and loss of function
- Some enzyme defects can be treated with enzyme substitution therapy
Lysosomal storage diseases are due to homozygous enzyme defects; they are rare, often clinically serious conditions. Out of the considerable variety of diseases, we will pick Gaucher disease as an example.
| 10.4.1 |
Biochemistry of Gaucher disease |
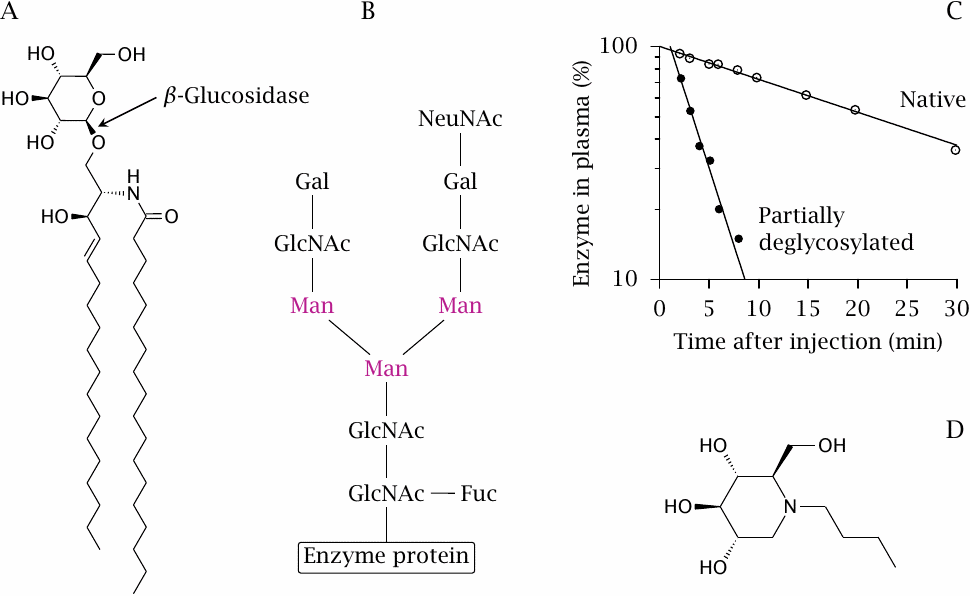
This slide summarizes the biochemical aspects of Gaucher disease. The gene defect concerns the enzyme lysosomal β-glucosidase or glucocerebrosidase, which cleaves the glucose moiety from glucocerebroside (glucosylceramide), a sphingolipid found in cell membranes (A). In Gaucher patients, the lipid accumulates in macrophages in the liver, spleen, and bone marrow. Liver and spleen become vastly enlarged and functionally deficient. Only a small fraction of the extra mass actually consists of glucosylceramide; the exact cause of the surplus enlargement is not understood.
β-Glucosidase is a glycoprotein; the structure of its glycosyl moiety is shown in panel B.73 Partial enzymatic removal of the oligosaccharide moiety up to the dotted line exposes mannose residues, which facilitate uptake of the enzyme by macrophages via a mannose-binding protein on the cell surface.
Panel C shows that the partially deglycosylated enzyme is removed from blood plasma much faster than the unmodified enzyme, which reflects the increased efficiency of uptake by macrophages.74 Replacement therapy with such enzyme preparations has very good clinical effectiveness and has become the standard treatment. Figure prepared from original data in [88].
Panel D shows the structure of miglustat, an inhibitor of glucocerebroside synthesis that is used in the treatment of Gaucher disease. Since glucocerebroside is an essential membrane constituent, inhibition of its synthesis cannot be complete, so this principle can only be used as an adjunct treatment.
| 10.5 |
Diabetes mellitus |
-
Lack of insulin activity due to
- destruction of pancreatic island β cell (type 1)
- loss of insulin sensitivity in peripheral organs (type 2)
- excessive levels of hormones antagonistic to insulin
- Blood glucose accumulates and causes acute and chronic pathology
- Treated with insulin substitution (type 1 and 2) and oral drugs (type 2)
Insulin is a peptide hormone that directs the utilization of dietary glucose. In most tissues, uptake of glucose by facilitated diffusion must be activated by insulin. In addition, insulin promotes the expression of enzymes that degrade glucose and convert it to fatty acids. The insulin receptor is a membrane-associated receptor tyrosine kinase. The release of insulin from the β cells in the pancreatic islets is induced by the blood level of glucose itself (see slide 6.6.2).
The destruction of the pancreatic islet cells in type I diabetes is due to an autoimmune reaction that is triggered by coxsackievirus infections. These infections are typically contracted at young age, which is the reason why type I diabetes is also known as juvenile diabetes.
Type II diabetes typically sets in during middle or advanced age; it is more common in overweight patients. The underlying mechanism of diabetes type II is still not well understood. In contrast to type I diabetes, the capacity of the β cells for producing insulin is preserved, and one therapeutic approach is to use drugs that amplify the secretion of insulin. In advanced stages, endogenous insulin production declines, and insulin substitution becomes necessary, as it is right from the beginning in type I diabetes.
Symptomatic diabetes often arises from an excess of glucocorticoid hormones, which may be due either to their use as drugs or to their formation by tumors in the adrenal gland. Excessive activity of thyroid or catecholamine hormones can also give rise to symptomatic diabetes.
| 10.5.1 |
Oral antidiabetic drugs |
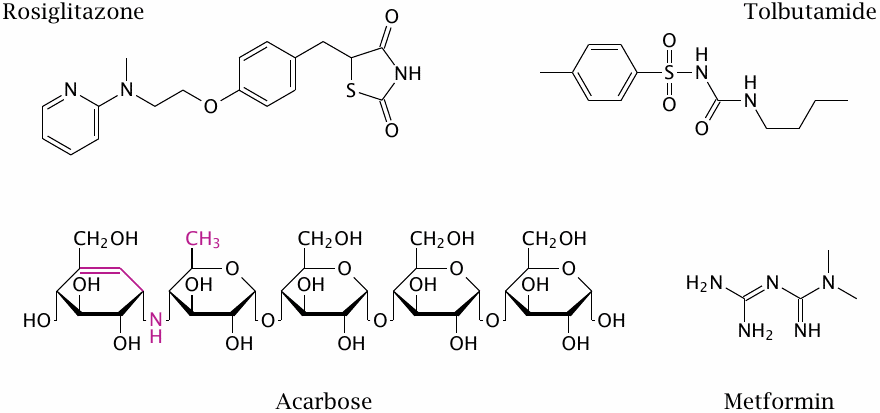
Oral antidiabetic drugs are a viable option only with type II diabetics in whom glucose regulation still works but is functionally inadequate. The therapeutic rationale is variously to slow glucose uptake, to increase insulin secretion, or to improve the diminished insulin sensitivity in peripheral tissues.
Acarbose is an inhibitor of intestinal maltase, the enzyme that cleaves maltose and other amylose digestion fragments to glucose. Since only monomeric glucose can be taken up from the intestinal lumen, maltase inhibition slows down glucose uptake, thereby reducing the peak load on glucose regulation.
The action mode of tolbutamide has already been discussed earlier (see slide 6.6.2). Rosiglitazone, a thiazolidinedione, reduces insulin resistance in type 2 diabetes. It is an agonist of the PPARγ, a nuclear hormone receptor that regulates the transcription of numerous enzymes and transporters involved in glucose and fat metabolism. but it has numerous side effects including the promotion of heart failure that have led to its withdrawal from the market in Europe. More recently, thiazolidinediones were found to specifically inhibit the mitochondrial pyruvate transporter [89,90]. This should reduce ATP and increase AMP levels in the cell. The next slide, which considers metformin, discusses how this might translate into an antidiabetic effect.
| 10.5.2 |
Hypothetical mode of action of metformin |
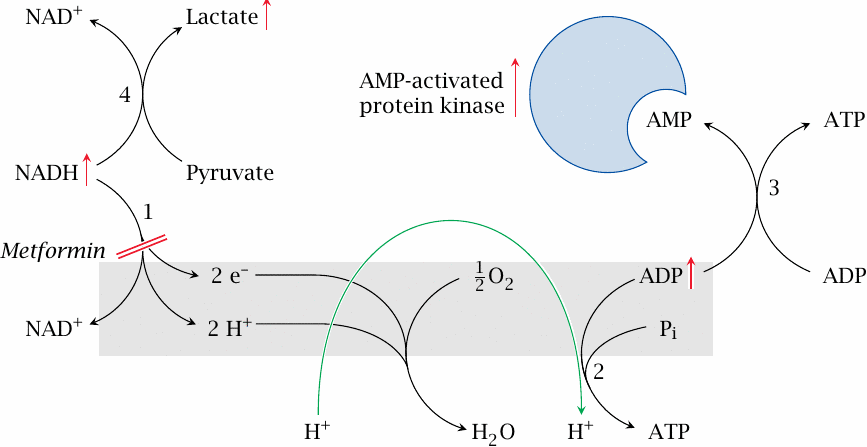
Metformin promotes activation of AMP-dependent protein kinase, which improves insulin-independent glucose utilization. This slide illustrates a hypothetical mechanism for this activation.
NADH dehydrogenase (1) is partially inhibited by metformin. This slows the entire respiratory chain and therefore the regeneration of ATP from ADP by ATP synthase (2). Adenylate kinase (3) converts ADP to AMP, which binds to AMP-activated protein kinase. The activated kinase stimulates glucose transport, even in the absence of insulin, as well as catabolic pathways. Another potentially relevant effect of AMP is the inhibition of adenylate cyclase [91], which would counteract the effect of glucagon and epinephrine, and so help to restore the balance between insulin and its antagonists.
Accumulated NADH promotes the reduction of pyruvate by lactate dehydrogenase (1). Depletion of pyruvate inhibits gluconeogenesis, and accumulation of lactate promotes lactate acidosis, which is a known side effect of metformin.
| 10.6 |
Atherosclerosis |
- Degenerative and inflammatory disease of the arteries
- Promoted by hypercholesterolemia, hypertension, diabetes, smoking
- Damaged arteries subject to chronic or acute obstruction, hemorrhage
- Most common cause of death, ahead of all cancers and leukemias combined
- Treatment strategies address cholesterol levels, blood pressure, blood coagulation
Atherosclerosis is not a purely metabolic disease; hypertension is another very important causal factor, and we have already noted several types of drugs that are used to treat it. Here, we will focus on the metabolic aspects of atherosclerosis and the related pharmacology.
| 10.6.1 |
Appearance of atherosclerotic lesions |
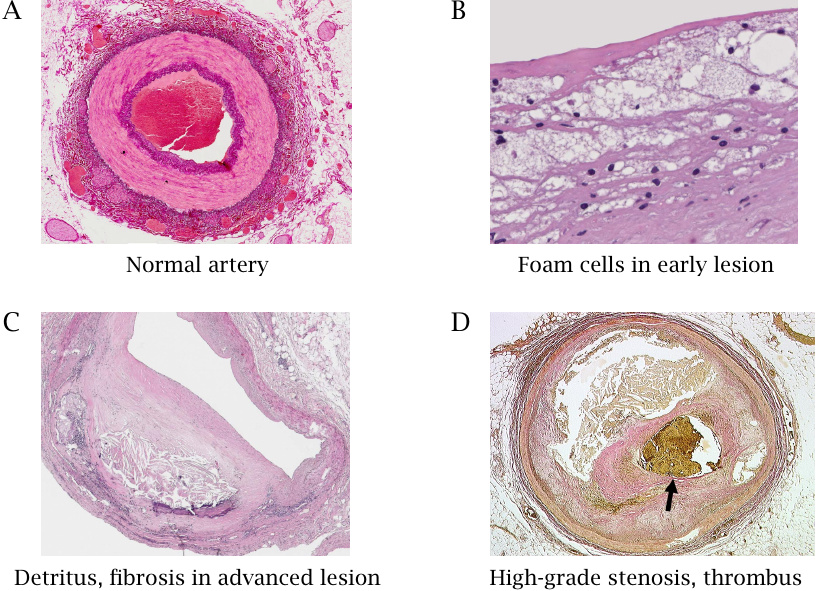
This slide shows cross sections of a normal artery (A) as well as those of arteries with atherosclerotic lesions in different stages of advancement (B-D). The normal artery displays inner and outer layers of connective tissue, stained in dark purple, as well as a strong intermediate muscular layer that shows up in a lighter tone. The endothelium is too thin to be discerned in this low power magnification view. The blood clot in the lumen is a post-mortem artifact.
Panel B shows a higher power view of an early lesion. The bubbly appearance is due to foam cells, which are macrophages stuffed chock-full with lipids. Panel C shows an advanced lesion with connective tissue proliferation and accumulation of detritus; the lumen of the blood vessel is considerably reduced. Panel D shows an artery that was already almost completely obliterated by a proliferating lesion that had spread around the entire circumference; the small residual lumen is blocked by an acutely formed thrombus (stained yellow-brown). Images reproduced with permission from pathorama.ch.
| 10.6.2 |
Development of an atherosclerotic lesion |
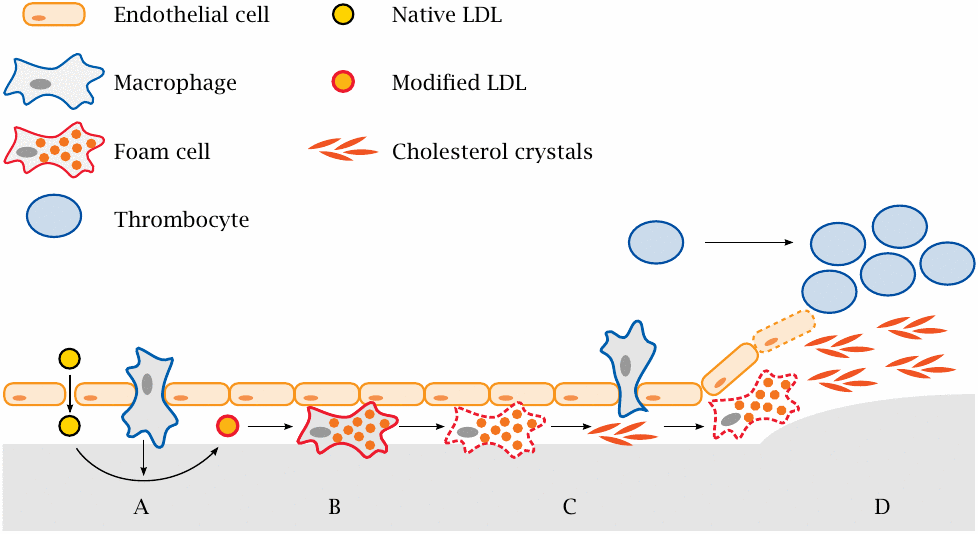
This slide summarizes the developmental stages of an atherosclerotic lesion. LDL is low density lipoprotein, a specific type of plasma lipoprotein particles that is enriched in cholesterol.
- (A)Small defects in the endothelium cause seepage of LDL into the subendothelial tissue, followed by transmigration of macrophages. Reactive oxygen species and enzymes released by macrophages modify the LDL.
- (B)Modified LDL is taken up by macrophages via scavenger receptors. Unlike the uptake of unmodified LDL via regular LDL receptors, the uptake mediated by scavenger receptors is not subject to feedback inhibition and thus causes lipid overload, which turns macrophages into foam cells.
- (C)Foam cells perish, disintegrate and release accumulated cholesterol, which forms crystalline deposits. These crystals activate new macrophages and cause them to release inflammatory cytokines that further incite and amplify the inflammation.
- (D)In an advanced lesion, cells in the muscular layer proliferate, progressively constricting the artery. When the endothelium that covers the lesion becomes eroded, thrombocytes and plasmatic coagulation factors are activated and initiate blood clotting, causing acute thrombus formation and obstruction.
As an alternative to thrombus formation, acute failure of a damaged artery can also occur through rupture. Approximately 20% of all cerebral infarctions are caused by rupture and hemorrhage rather than thrombotic occlusion.
| 10.6.3 |
Transport and metabolism of cholesterol |
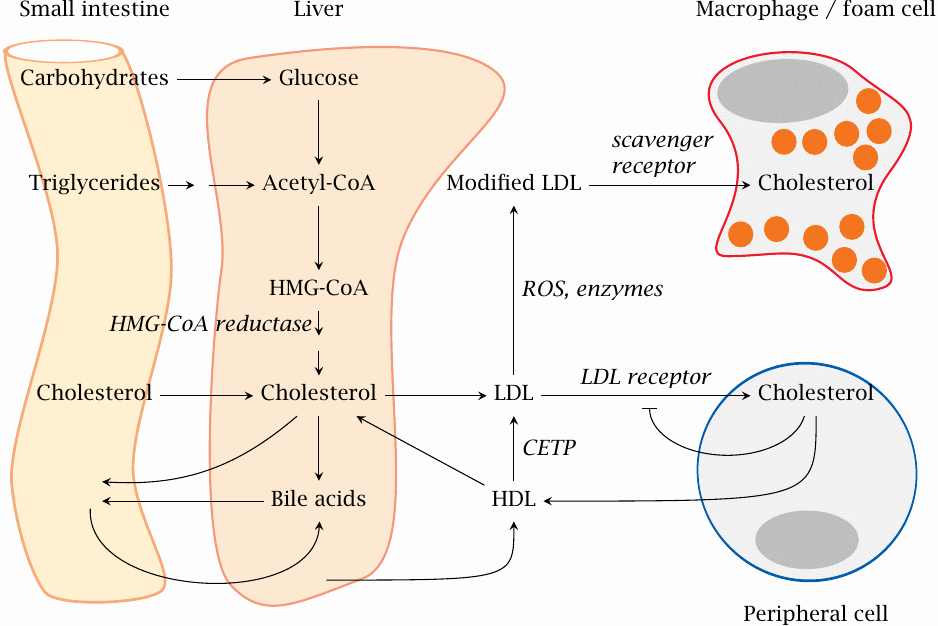
Cholesterol is the key lipid that accumulates in atherosclerotic lesions and sustains the inflammation and subsequent degeneration. Lowering cholesterol is one of the most important goals in the treatment of atherosclerosis. To devise proper strategies, we first need to understand how cholesterol is formed, transported and eliminated.
The pool of cholesterol in the liver is replenished by uptake of dietary cholesterol and by synthesis from other foodstuffs via acetyl-CoA and hydroxymethylglutaryl-CoA (HMG-CoA). It is transported from the liver to peripheral tissues packaged into low density lipoprotein (LDL) particles. Cells in the peripheral organs take it up by endocytosis via LDL receptors (LDL-R). Uptake via this receptor is subject to feedback inhibition by intracellular cholesterol.
Excess cholesterol is returned from peripheral cells to the liver via HDL, but on the way some of it is transferred back from HDL to LDL by cholesterol-ester transfer protein (CETP). The liver also uses cholesterol to synthesize bile acids, which are secreted into the bile along with some excess cholesterol. Bile acids undergo reuptake at the end of the small intestine and return to the liver.
As discussed above, within atherosclerotic lesions, LDL undergoes modification by reactive oxygen species (ROS) or degradative enzymes. Enzymatically or chemically modified LDL enters macrophages via scavenger receptors, overloads them with cholesterol and turns them into foam cells.
| 10.6.4 |
Intestinal cholesterol uptake |
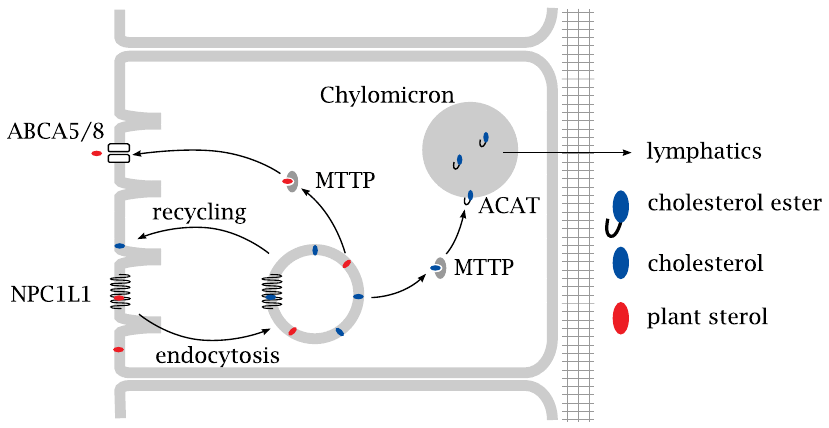
Uptake of dietary cholesterol into the cells of the intestinal mucous membrane occurs through endocytosis and is controlled by the membrane protein NPC1L1. From the ingested vesicles, cholesterol is shuttled to the endoplasmatic reticulum by the microsomal triglyceride transport protein (MTTP; obviously a bit of a misnomer). In the ER, cholesterol is esterified by acyl-CoA-cholesteryl-acyltransferase (ACAT) and incorporated into nascent chylomicrons, which are large lipoprotein particles that also carry triacylglycerol.
The chylomicrons are released on the basolateral side and make their way to the liver via the lymphatics and the blood circulation. Plant sterols such as sitosterol will also be taken up by endocytosis but are ejected back into the intestinal lumen through an ABC5/8, an ABC transporter.
| 10.6.5 |
Inhibitors of intestinal cholesterol uptake |
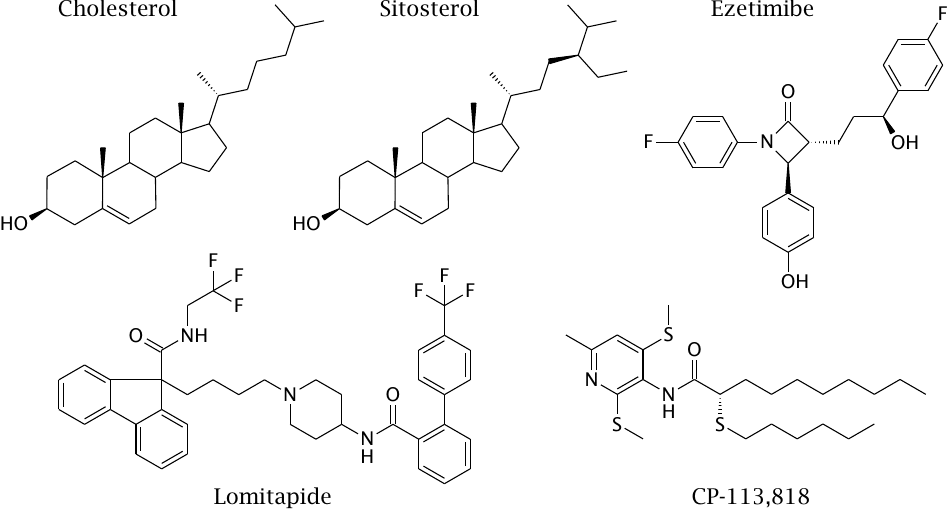
Inhibition of intestinal cholesterol uptake is, on its own, not sufficient for controlling cholesterol, but it is useful in combination with inhibitors of cholesterol synthesis (see below).
Sitosterol is a natural plant sterol that competes with cholesterol for uptake into the intestinal cell by endocytosis, but is then ejected back into the gut (see previous slide). Ezetimibe is an inhibitor of NPC1L1. Lomitapide and CP-113,818 are experimental inhibitors of MTTP and of ACAT, respectively.
| 10.6.6 |
Inhibition of HMG-CoA reductase with statins |
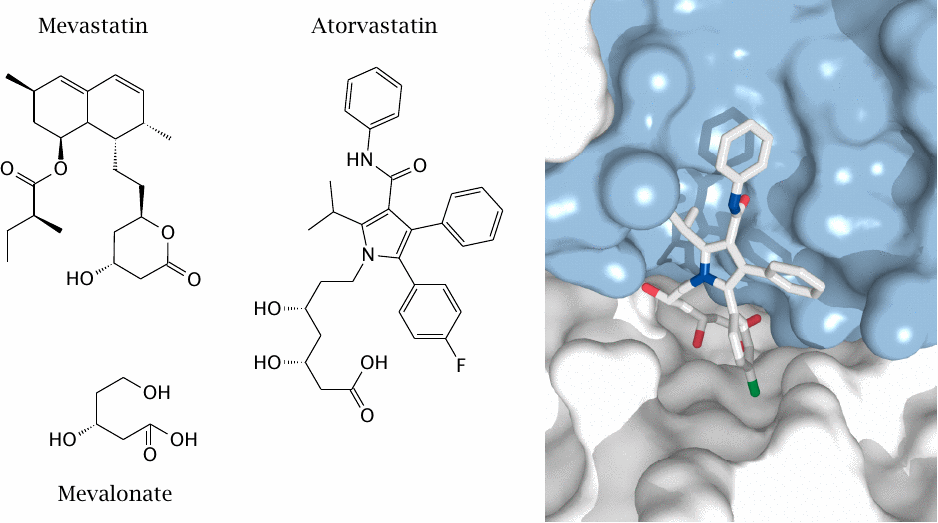
The key regulated step in cholesterol biosynthesis is catalyzed by hydroxymethylglutaryl-CoA (HMG-CoA) reductase, which converts its substrate to mevalonate. This enzyme is targeted by the so-called “statin” drugs.
Mevastatin, a natural antibiotic, was the first such drug to be isolated. Atorvastatin is a synthetic inhibitor; it is shown on the right within the active site of HMG-CoA reductase. Both inhibitors contain a moiety that resembles mevalonate.
| 10.6.7 |
Overview of blood coagulation |
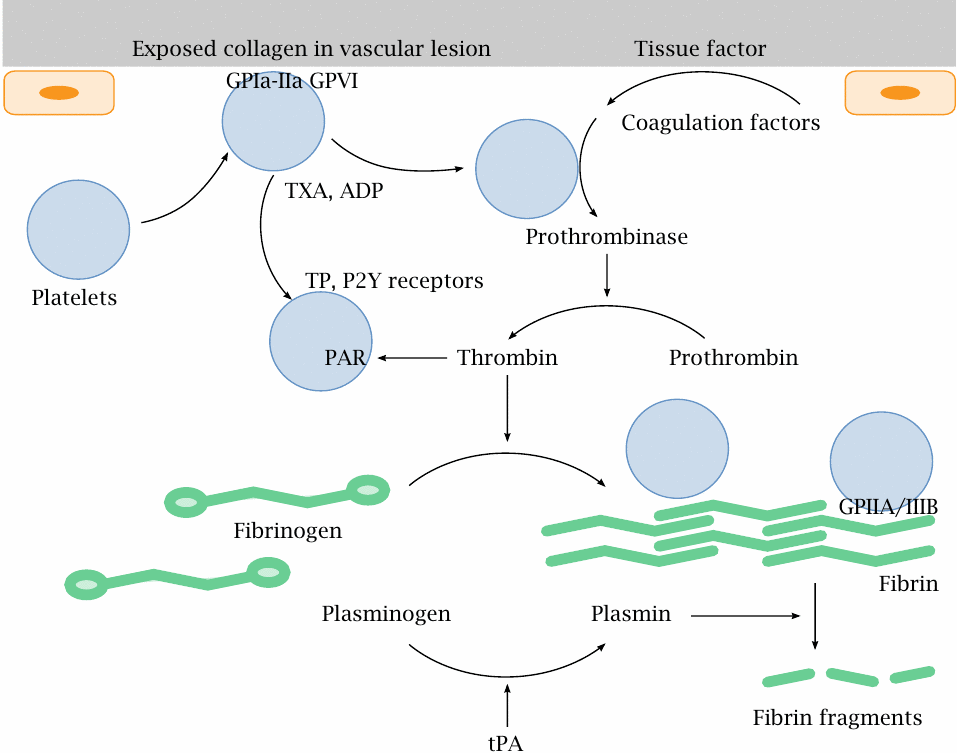
While reduction of cholesterol and blood pressure help to prevent the progress of atherosclerosis, they cannot do much to revert manifest vascular lesions. In patients with advanced atherosclerosis, it therefore becomes important to lower the risk of thrombus formation atop those lesions. This is done by partial inhibition of blood coagulation. Such treatment must be used with care; if coagulation is inhibited too strongly, the increase in the risk of hemorrhage may exceed the reduction in the risk of thrombotic infarction.
Blood coagulation is initiated by defects in the vascular endothelium, which expose collagen fibers. Platelets (thrombocytes) bind to collagen via their collagen receptors (GP1a-IIa, GPVI) and begin to secrete ADP and thromboxanes. These mediators activate further platelets through purine (P2Y) and thromboxane (TP) receptors, respectively. Plasmatic coagulation factors sequentially activate one another by proteolysis at the site of the lesion and on the surface of activated platelets; this culminates in the activation of thrombin (factor II). Thrombin then cleaves fibrinogen to fibrin, which forms the clot. It also cleaves protease-activated receptors (PAR) on platelets and so further amplifies platelet activation. Platelets adhere to fibrinogen or fibrin via GPIIA/IIIA receptors.
Both plasmatic coagulation and platelets can be inhibited with drug therapy. On platelets, all of the receptors discussed above can be targeted. We have already seen ticlopidine, which inhibits P2Y receptors (slide 6.16.1), as well as ramatroban, which blocks both the synthesis and receptors of thromboxane A2 (slide 9.6). Low-dose aspirin therapy has been discussed as well (slide 9.5.1).
The fibrin clot is ultimately dissolved by plasmin, which is proteolytically released from plasminogen by tissue plasminogen activator (tPA). The latter enzyme is used to dissolve blood clots and so restore organ perfusion in acute cases of myocardial infarction and stroke. The related protease urokinase (see slide 1.1.2) can be used for the same purpose.
| 10.6.8 |
Inhibition of plasmatic blood coagulation with warfarin |
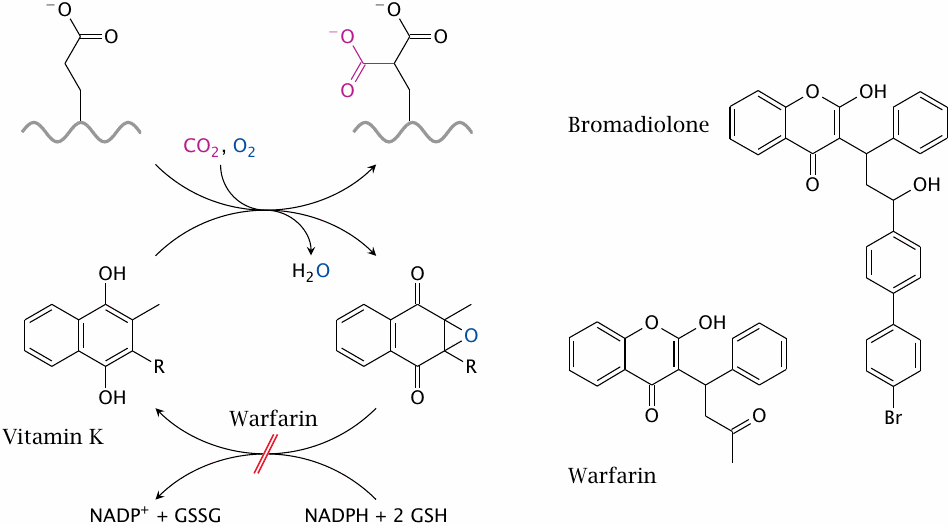
Plasmatic coagulation can be inhibited by antimetabolites of vitamin K. Several coagulation factors (II, VII, IX, and X) require posttranslational modification of glutamate residues to γ-carboxyglutamate. The modified residue enables these proteins to chelate calcium, which in turn lets them bind to the surfaces of activated thrombocytes. The enzyme that carries out the modification, γ-glutamyl carboxylase, requires reduced vitamin K as a cosubstrate and converts it to an epoxide. The latter is reduced again by vitamin K epoxide reductase, which is inhibited by the drug warfarin. (In the structures of vitamin K and its epoxide, R represents a side chain containing several isoprenoid residues.)
Complete inhibition of vitamin K reductase is, of course, deleterious, and indeed is used to kill rats with poisons such as bromadiolone. Therefore, this type of drug has a very low therapeutic index and requires careful monitoring. Thrombocyte aggregation inhibitors are less precarious and are usually preferable in clinical practice.