| 11 |
Chemotherapy of infectious diseases |
| 11.1 |
Introduction |
In the treatment of infections, the goal is to kill the microbes, or at least to disrupt their ability to propagate, so that the immune system can get the better of them. While this is simple in principle, the difficulties in practice are twofold. Firstly, the drugs must kill the microbes but spare the host; this is the principle of selective toxicity. Secondly, microbes tend to develop resistance to drugs that are initially effective. The problem of resistance is both grave and unavoidable, since microbes are subject to genetic variation, and each drug use, medically justified or not, will select for more resistant variants. Therefore, antimicrobial drug discovery may have to continue in perpetuity.
| 11.2 |
Diversity of infectious pathogens |
- Bacteria
- Fungi
-
Parasites—eukaryotes other than fungi
- Protozoa—unicellular
- Metazoa—multicellular
- Viruses
Among the parasites, the category “protozoa” is a bit of a historical relic. When infectious agents were first identified, the genetic and biochemical relationships between them were not understood, and newly discovered pathogens were classified somewhat haphazardly and mostly according to appearance and shape. The term “protozoa” means “primitive animals”. However, as shown in the next slide, the protozoa are rather distantly related to humans and other real animals, and even to one another.
The large evolutionary distance between protozoa and humans corresponds to significant differences in cellular biochemistry. Some of these biochemical differences can be exploited to achieve selective toxicity in chemotherapy.
| 11.2.1 |
The tree of life, slightly pruned |
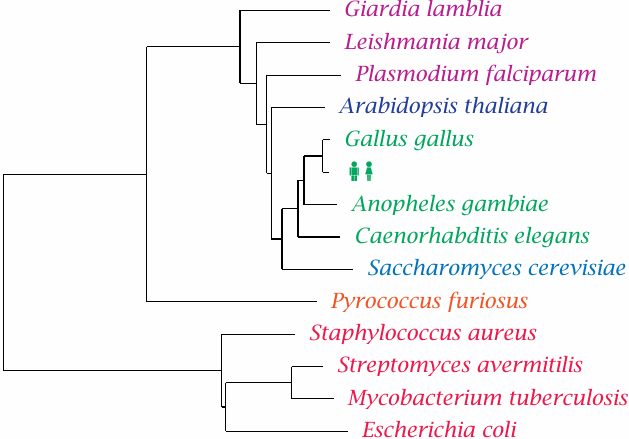
This phylogenetic tree shows Homo sapiens in the company of some pathogens and reference organisms. Arabidopsis is a plant, and Saccharomyces is a yeast; both are more closely related to humans than are Plasmodium, Leishmania and other single-celled eukaryotic parasites that have been lumped in with the protozoa. The nematode Caenorhabditis elegans is not a pathogen itself but is included as a proxy for human worm pathogens such as Wuchereria bancrofti or Ascaris lumbricoides.
Pyrococcus furiosus75 belongs to the Archaebacteria. These organisms have a prokaryotic cell structure but are phylogenetically closer to us than to the Eubacteria, to which all pathogenic bacteria belong. Among these, Staphylococcus aureus represents the Gram-positives, whereas Escherichia coli represents the Gram-negatives. These two major groups have different cell wall structures (see slide 11.4.1) that differ in their permissiveness both for the dyes used in the Gram stain procedure and for antibiotics. Mycobacteria such as Mycobacterium tuberculosis, which are genetically almost equidistant from both, have another distinct cell wall architecture that is particularly hard to penetrate for antibiotics.
The cell wall structure of the streptomycetes resembles that of the Gram-positives, but they are genetically closer to the mycobacteria. They occasionally occur as pathogens but are most notable as purveyors of antibiotics; the majority of all known natural antibiotics is produced by member species of Streptomyces or related genera.
| 11.2.2 |
Drug targets for antimicrobial therapy |
-
Macromolecules that occur in the cells of the pathogen but not within the human
host. Examples:
- the bacterial cell wall (penicillin)
- de novo synthesis of folic acid (sulfonamides)
-
Macromolecules that occur in both humans and the pathogen but are structurally
divergent. Examples:
- ribosomes (chloramphenicol)
- dihydrofolate reductase (trimethoprim)
- DNA topoisomerase (ciprofloxacin)
This list of examples shows that both pathogen-specific drug targets and those that are shared but divergent are important and viable in clinical practice.
In recent years, the genomes of many pathogens have been sequenced and searched for genes that are essential for the life of the pathogens, and at the same time do not have counterparts in the human genome. The products of such genes should indeed make good candidate drug targets. However, considering the proven value of many drugs that act on gene products that do have homologues in host cells, we should not exclude the latter from our search for novel targets.
Both types of drug targets—those with and those without homologues in the host cell—tend to be more abundant in pathogens with greater evolutionary distance from humans. Thus, the number of suitable targets is smaller in protozoal parasites than in bacteria, and yet smaller in pathogenic fungi.
| 11.2.3 |
Structures of folic acid and of three inhibitors of dihydrofolate reductase |
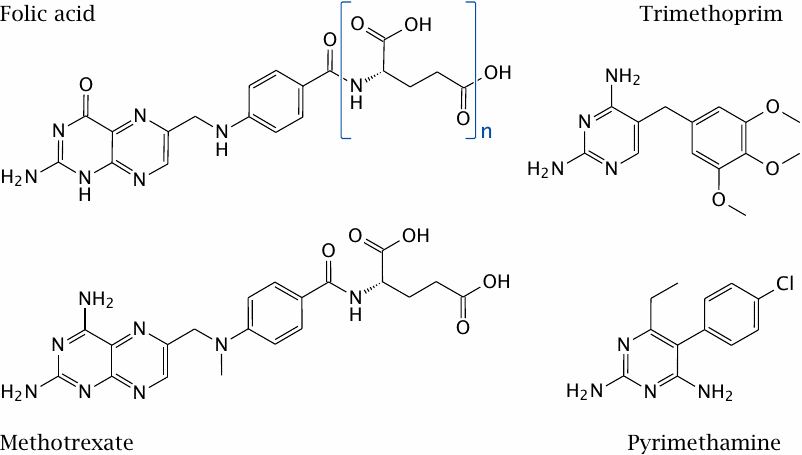
Dihydrofolate reductase is a good example of a drug target that is found in both host and pathogen. It is essential in all organisms for sustaining the folic acid-mediated transfer of single-carbon groups in biosynthetic reactions. Nevertheless, the enzyme molecules found in various organisms are sufficiently different to allow their selective inhibition with various chemotherapeutic drugs.
Trimethoprim inhibits the bacterial enzyme. It acts synergistically with sulfonamides, which inhibit the bacterial de novo synthesis of folic acid (see slide 1.3.3). Trimethoprim is typically used in combination with a sulfonamide.
Pyrimethamine inhibits the enzymes of protozoans such as Toxoplasma gondii and Plasmodium falciparum. The human enzyme is not significantly inhibited by either but is inhibited by methotrexate, which is used in anticancer and immunosuppressive therapy.
| 11.2.4 |
Microbial resistance mechanisms |
-
Mechanisms affecting the target:
- Structural alteration / mutation
- Compensatory overexpression
-
Mechanisms affecting the drug:
- Reduced uptake
- Active extrusion
- Enzymatic inactivation
Resistance to chemotherapy occurs with all kinds of pathogenic microbes. While it poses obvious and significant practical problems, it is also a fascinating microcosm of Darwinian evolution through mutation and selection. Larger organisms mostly evolve too slowly for us to observe; with drug resistance in microbes, we get a time-lapse view of evolutionary adaptation. This is due to both the short generation time of microbes and the rigorous nature of the selection.
This slide lists the principal mechanisms of resistance that apply to all classes of pathogenic microbes. We will see specific examples for most of these mechanisms in the following sections.
| 11.3 |
Overview of antibacterial chemotherapy |
-
Targets
- Cell wall
- Ribosomes
- Enzymes related to cell division
- Intermediate metabolism
-
Antibiotic resistance
- Bacteria have short generation times—fast de novo evolution of resistance
- Resistance genes exist in producers of antibiotics—can spread to pathogenic bacteria by gene transfer
We will start our exploration of chemotherapeutic agents with those that act on bacteria, which are the most common class of pathogens.
Most antibacterial drugs are antibiotics, that is, natural compounds isolated from other microbes, or derivatives thereof. While penicillin was famously isolated from a mold, most antibiotics—for example, tetracyclins, aminoglycosides like streptomycin, and macrolides like erythromycin—are actually produced by Streptomyces species or related soil bacteria. Since these producer bacteria must be resistant to their own poisons, it follows that mechanisms and genes for bacterial resistance must exist for any of these natural antibiotics. Such genes may migrate to clinical pathogens and spread among them if we apply the proper selection pressure through the medical use and misuse of those antibiotics.76
Some antibacterial compounds are indeed fully synthetic; we have already seen sulfonamides and trimethoprim as examples. With these agents, resistance genes may not exist a priori; however, resistance often emerges through spontaneous mutations that sharpen the target enzymes’ ability to discriminate between the drugs and the proper substrates.
With both natural and synthetic antibiotics, an important strategy to prevent, or at least delay, the emergence of resistance is combination therapy. When several drugs are combined that each alone are able to kill the pathogen, and each of which addresses a different target, the pathogen would have to simultaneously modify all targets in order to survive. With increasing number of agents simultaneously applied, this rapidly becomes unlikely.77
| 11.3.1 |
Gene transfer mechanisms in bacteria |
- Transformation: cellular uptake of naked DNA
- Conjugation: plasmid-encoded active transfer between bacterial cells
- Transduction: gene transfer mediated by bacteriophages
- Transposons: transfer of genes between carrier DNA molecules (chromosomes, plasmids)
Transformation, conjugation and transduction all work within and also between bacterial species.78 Transfer between species is important in the migration of resistance genes from producer strains, or other soil bacteria that are naturally exposed and have developed resistance to a given antibiotic, to pathogenic bacteria.
Resistance genes are often carried by mobile genetic elements called transposons, which encode enzymes with endonuclease and ligase activity that allow them to excise themselves from one host DNA molecule and then join another. Given the right selection conditions, multiple resistance transposons may wind up on a single plasmid molecule. Such a plasmid will then confer resistance to several unrelated antibiotics all at once, and the use of any single one of these drugs will cause this multiple resistance to spread further. The first such multiresistance plasmids were observed in the late 1950s, only about ten years after the start of the antibiotic era.
| 11.4 |
Antibiotics and the bacterial cell wall |
In contrast to human cells, which are simply delimited by their cytoplasmic membranes, most bacteria have cell walls that consist of one or more protective layers stacked on top of their cytoplasmic membranes. While these cell walls may protect the bacteria from antibiotics, they also provide targets for chemotherapy. Note that some bacteria—mycoplasmas, and the vegetative forms of rickettsias and chlamydias—have no cell wall at all and therefore are not susceptible to the agents discussed in this section.
| 11.4.1 |
Bacterial cell wall structure |
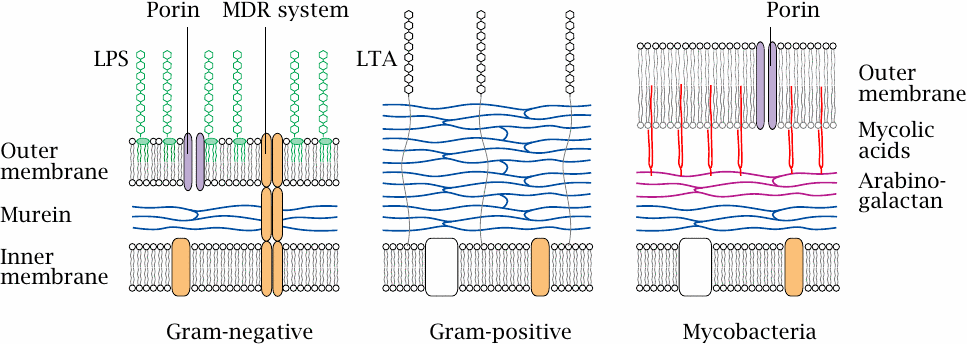
The innermost layer of a bacterial cell wall consists of murein or peptidoglycan, a meshwork of polysaccharide strands crosslinked by oligopeptides. Gram-negative bacteria have a comparatively thin peptidoglycan layer (blue) that is surrounded and protected by an outer membrane. The outer leaflet of this lipid membrane consists mostly of lipopolysaccharide (LPS, green), which is also known as endotoxin.79 Porins in the outer membrane facilitate diffusion of small polar solutes. Multidrug resistance (MDR) proteins that extrude antibiotics from the cell may be located in the cytoplasmic membrane alone or span both membranes.80
Gram-positive bacteria lack an outer membrane but have a much thicker murein layer, which is decorated with lipoteichoic acids (LTA). The lack of an outer membrane makes them more amenable to penicillin (see slide 1.3.10) and many other antibiotics.81
In mycobacteria, the murein layer is surrounded by arabinogalactan polysaccharide, to which branched, long-chain fatty acids (mycolic acids, red) are attached. The mycolic acids act as anchors for a particularly thick, wax-like and impenetrable outer membrane. Because of their sturdy cell wall,82 mycobacteria have always been among the most difficult microbes to treat—although decades of selection have bred some real champions of resistance among the Gram-positives and Gram-negatives also.
| 11.4.2 |
Action mechanism of isoniazid (INH) |
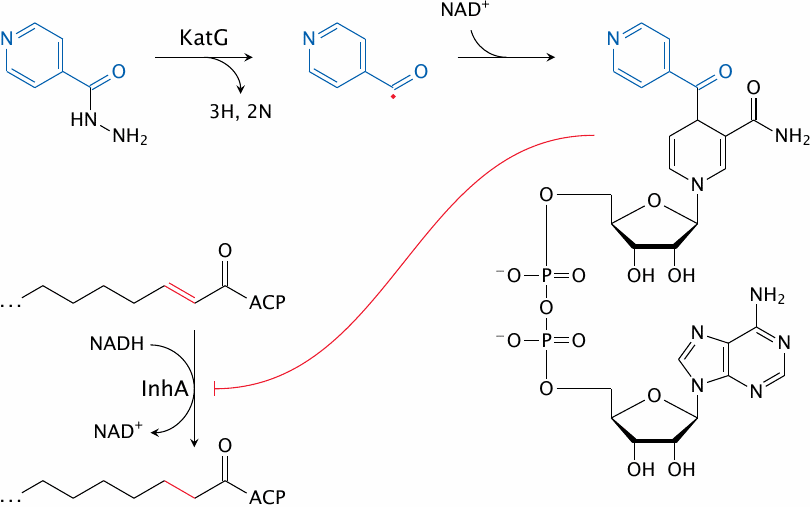
Isoniazid is effective against the pathogen Mycobacterium tuberculosis. It inhibits the synthesis of mycolic acids, the long-chain fatty acids that are characteristic and essential components of the mycobacterial cell wall. This inhibition arises in a rather unique manner.
Inside the mycobacterial cell, INH is activated by the oxidative enzyme KatG83 to a radical form. This radical then reacts with NAD+. The adduct inhibits InhA, an enoyl-CoA reductase involved in mycolic acid synthesis.
| 11.4.3 |
Outline of bacterial murein synthesis |
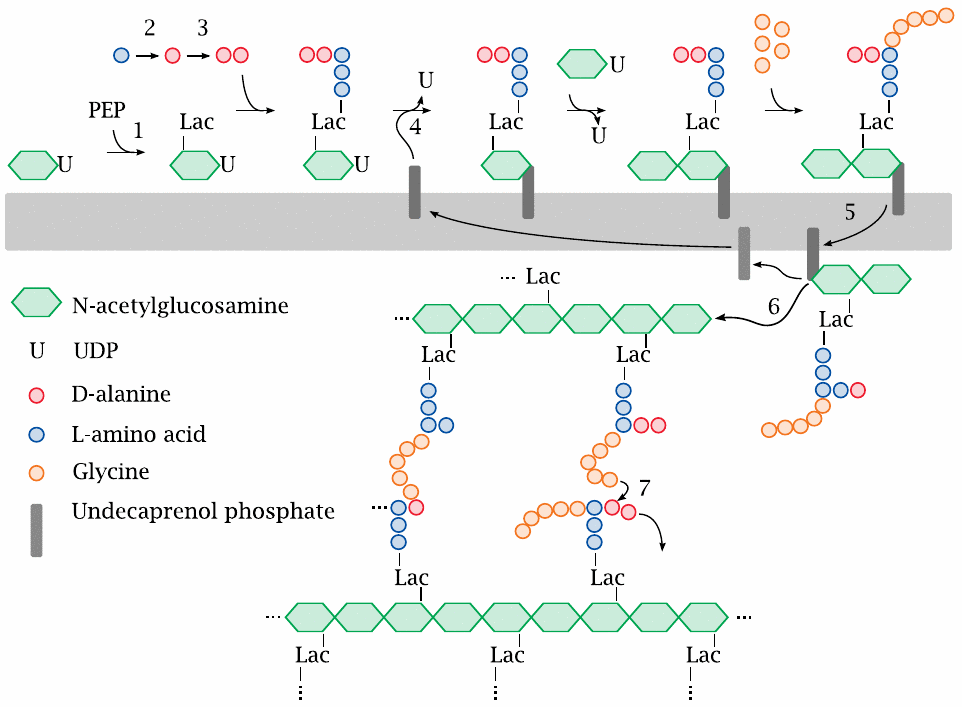
Murein, or peptidoglycan, is found in the cell walls of Gram-positives, Gram-negatives, and mycobacteria. Inhibitors of various enzymes in the murein synthesis pathway are active against member species of all three classes.
The synthetic pathway involves the following stages: Phosphoenolpyruvate (PEP) supplies a lactate residue (Lac) that is attached to N-acetylglucosamine, which yields N-acetyl-muramic acid (1). Onto the latter, a pentapeptide is built in a series of ATP-activated reactions. The free end of this peptide contains two d-alanine residues that are supplied by alanine racemase (2) and d-alanine ligase (3). This nascent building block is transferred to the lipid carrier undecaprenol phosphate (4) and subsequently extended by another molecule of N-acetylglucosamine and five glycine residues.
The completed precursor molecule, named lipid II, is flipped across the cytoplasmic membrane (5). The glycopeptide moiety is transferred from lipid II to a growing murein strand in the transglycosylase reaction (6). The final transpeptidase reaction (7) cross-links the new subunit to an adjacent murein strand, releasing the terminal d-alanine residue.
The transglycosylase and transpeptidase activities are both located on the same enzyme protein, variously referred to as muramyl-transpeptidase or penicillin-binding protein (PBP). Most bacterial species have several PBP subtypes that may differ in susceptibility to penicillins and related antibiotics.
| 11.4.4 |
Fosfomycin mimics both phosphoenolpyruvate and glycerophosphate |
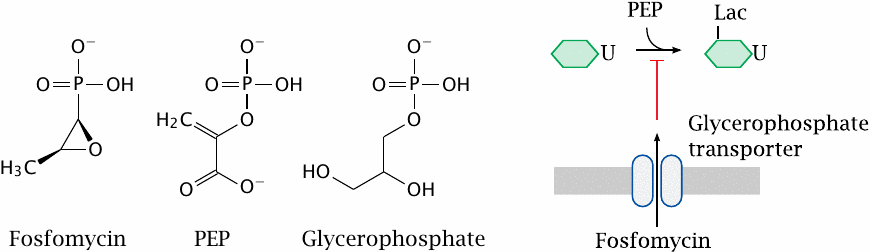
Fosfomycin is an antimetabolite of phosphoenolpyruvate (PEP) in the first step of murein precursor synthesis. Considering its structure, it may not surprise you to learn that the reaction with the target enzyme is covalent and involves the thiol group of a cysteine residue [93].
For uptake across the cytoplasmic membrane, fosfomycin piggybacks on a transport protein that mediates the uptake of glycerophosphate. This transporter is not essential for the bacterial cell; therefore, mutations that inactivate the transporter tend to cause rapid development of resistance under therapy. Fosfomycin can therefore only be used in combination with other antibiotics that are less prone to rapid evolution of resistance.
| 11.4.5 |
Cycloserine inhibits alanine racemase and d-alanine ligase |
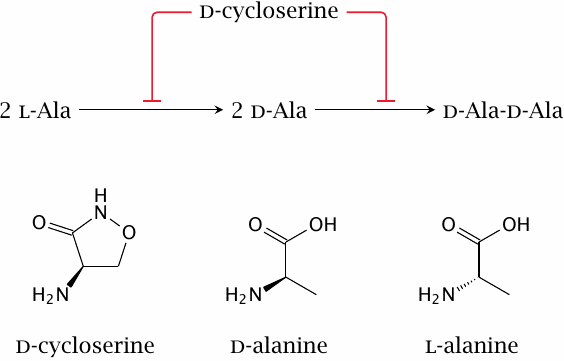
d-cycloserine is an antimetabolite of d-alanine in both the alanine racemase and the d-alanine ligase reactions. It is used mostly against mycobacterial infections, though active in principle against other types of bacteria also.
Interestingly, d-cycloserine is also a partial agonist at the sole glycine-specific subunit of the NMDA-type glutamate receptor (see section 6.11). As such, it has been tried therapeutically in various neurological and psychiatric conditions [94].
| 11.4.6 |
The lipopeptide laspartomycin sequesters undecaprenol phosphate |
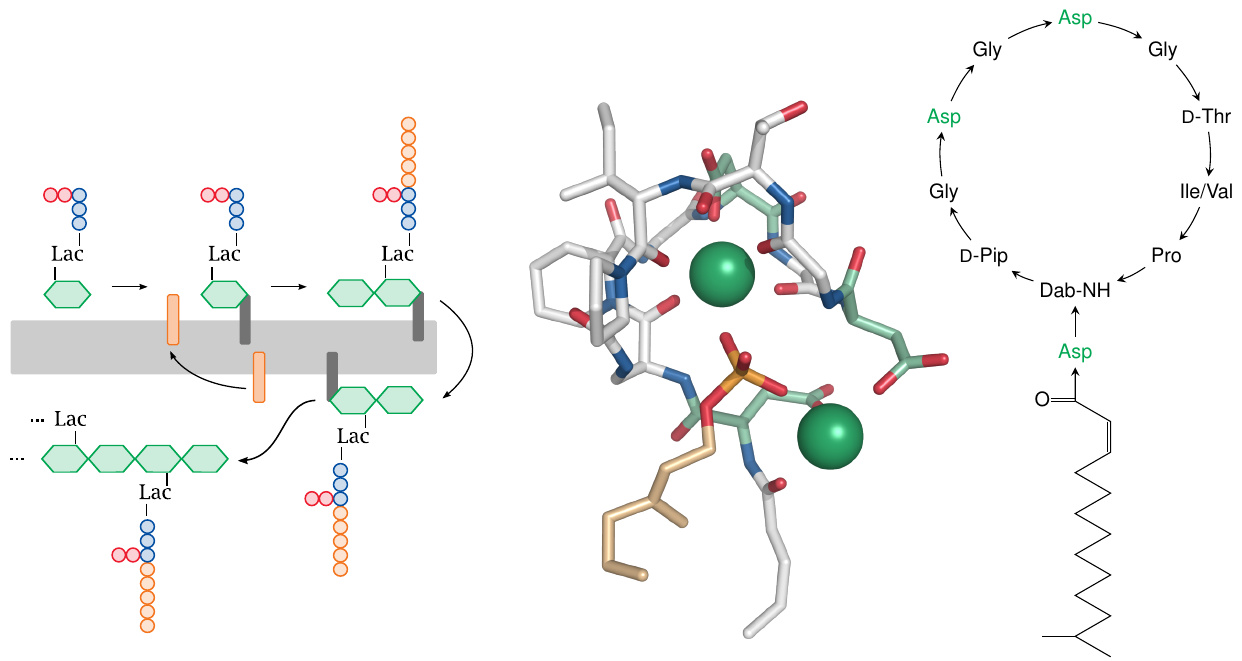
The acidic aspartate side chains participate in the binding of two calcium ions (green balls in the structure), which mediate binding to the likewise acidic phosphate headgroup of the undecaprenol carrier. Structure rendered from 5O0Z.pdb.
While mechanistically interesting, this antibiotic cannot be used in humans, at least not systemically, since it also interferes with the dolichol phosphate-dependent posttranslational glycosylation of proteins and therefore is toxic.
| 11.4.7 |
β-Lactam antibiotics resemble the substrate of the transpeptidase reaction |
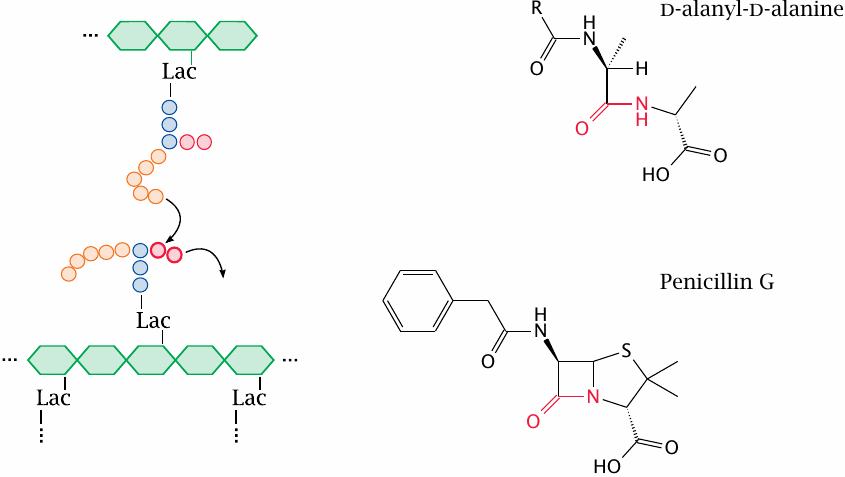
We now skip ahead to the last step of murein synthesis, that is, the transpeptidase reaction that crosslinks the assembled linear strands of murein and thereby confers mechanical stability to the cell wall.84
β-Lactam antibiotics, which inhibit the transpeptidase reaction, are the most widely used class of antibiotics. The class comprises penicillins, cephalosporins and a few other variants. Penicillin G, which is shown here, was the first β-lactam antibiotic to be introduced into clinical practice.
| 11.4.8 |
Reactions of penicillin G with transpeptidase and β-lactamase |
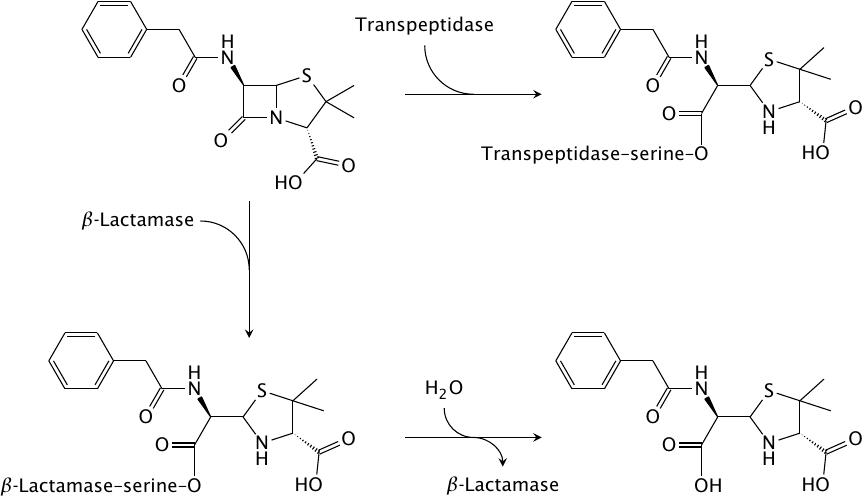
The shared structural feature that gave the class its name is also at the heart of its antibacterial action. The sterically strained four-membered β-lactam ring is spring-loaded like a mousetrap and readily reacts with the catalytic serine residue of the transpeptidase enzyme. The enzyme is unable to free itself from the covalent modification and remains irreversibly inactivated.
The most prevalent mechanism of bacterial resistance to β-lactams is enzymatic inactivation by β-lactamases. These enzymes fall into two major functional classes. Like transpeptidase, the first type of β-lactamase also contains a catalytic serine. However, in contrast to transpeptidase, the β-lactamase regenerates its free serine residue through hydrolysis, and it thus can successively degrade a large number of antibiotic molecules. Some enzymes in this class can be inhibited with β-lactamase inhibitors.
The second class of β-lactamases comprises metalloenzymes. In contrast to the serine β-lactamases, these enzymes do not form covalent intermediates, and no clinically useful inhibitors are currently available. Gram-negative organisms with metallo-β-lactamases have become a major problem worldwide. The Dmitrienko group in the UW chemistry department is working on the synthesis and characterization of such inhibitors [95].
| 11.4.9 |
Structures of β-lactam antibiotics (1) |
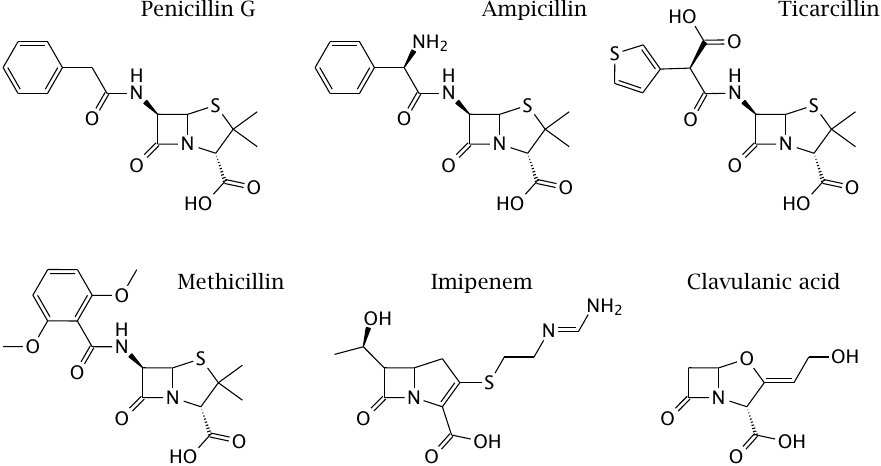
The first β-lactam to be used clinically, penicillin G, is active almost exclusively against Gram-positive bacteria, since it fails to penetrate the outer membrane of most Gram-negatives (see slide 1.3.10). Other β-lactams that were developed later have progressively extended the spectrum to include most Gram-negatives and even some mycobacteria. Some of these derivatives are also, to varying degrees, protected from inactivation by bacterial β-lactamases.
Penicillin G itself is readily cleaved by β-lactamases, and while it was active on almost all strains of Staphylococcus aureus when first introduced, more than 90% of all clinical isolates of this pathogen now possess β-lactamases that render them resistant.
To counter this resistance mechanism, semisynthetic drugs such as methicillin were developed, which escape cleavage by the enzyme through steric obstruction. This obstruction also renders such drugs about ten times less active against the muramyl-transpeptidase target, but because of the generally very high therapeutic index of the penicillins, this does not compromise their clinical utility. However, methicillin-resistant S. aureus strains (MRSA) have emerged and become increasingly widespread. Through horizontal gene transfer from another staphylococcal species, these strains have acquired a peculiar variant of the transpeptidase that no longer binds methicillin and related compounds. MRSA are also resistant to most other β-lactams; they are, however, susceptible to some new cephalosporins such as ceftobiprole (see next slide).
Ampicillin was the first penicillin derivative with useful activity against Gram-negatives such as Escherichia coli, and ticarcillin the first one to be active against Pseudomonas. Both are susceptible to now widespread β-lactamases, but they can still be used when combined with β-lactamase inhibitors such as tazobactam or clavulanic acid (see below).
Imipenem is an erstwhile “wonder drug” that killed just about anything when it first appeared in the market. It is intrinsically quite resistant to cleavage by serine-type enzymes but is cleaved by the increasingly common metalloenzyme β-lactamases. Imipenem lacks the sulfur atom in the ring and is therefore referred to as a carbapenem. Such structures were first observed among the thienamycins, a class of β-lactams produced by certain Streptomyces strains.
| 11.4.10 |
Structures of β-lactam antibiotics (2) |
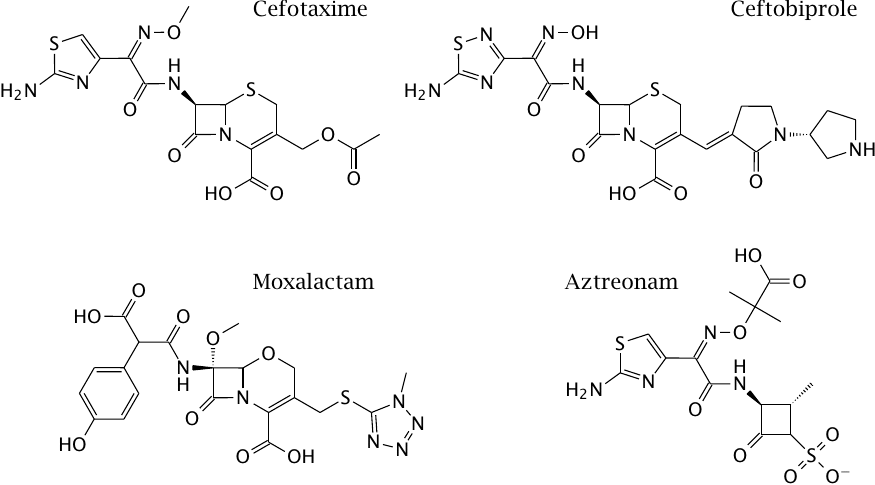
Cefotaxime and ceftobiprole are semisynthetic cephalosporins. Natural cephalosporins are produced by fungal species that belong to the genus Cephalosporium. Cephalosporins differ from the penicillins in the structure of the central nucleus. The cephalosporin nucleus is amenable to semisynthetic derivatization and variation in two positions. Like imipenem, many cephalosporins are quite resistant to serine β-lactamases but are susceptible to metalloenzymes. Cefotaxime has broad activity against both Gram-negative and Gram-positive bacteria; the more recently introduced ceftobiprole has the added bonus of being active against MRSA.
Moxalactam is an interesting cephalosporin analog with very good activity against some difficult Gram-negative pathogens, but it was retired due to interference with the plasmatic blood coagulation cascade. Aztreonam is a monobactam, an unusual, fully synthetic β-lactam antibiotic with a single ring structure. It has strong activity against Pseudomonas species.
| 11.4.11 |
Inactivation of SHV-1 β-lactamase by clavulanic acid |
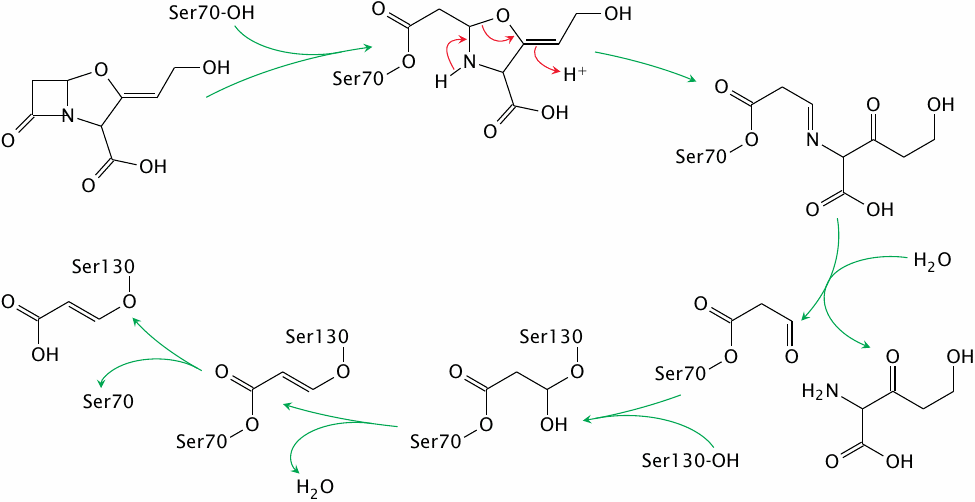
Clavulanic acid is a natural β-lactam antibiotic. Its intrinsic antibacterial activity is weak, but it is a suicide inhibitor of serine-type β-lactamases and as such can be used in combination with other, more active but β-lactamase-susceptible compounds such as ampicillin or ticarcillin.
The initial acylation of the catalytic serine 70 residue resembles the reaction of the enzyme with other β-lactam antibiotics. However, with clavulanic acid, this initial reaction is followed by a sequence of reactions that traps a second serine residue in the active site. The modification of serine 130 means that the active site remains blocked even after serine 70 is freed by hydrolysis.
Resistance to clavulanic acid can arise through mutation of serine 130 to glycine, which does not compromise the catalytic activity of the β-lactamase on penicillin derivatives. Reaction scheme simplified after [96].
| 11.4.12 |
Vancomycin sequesters the substrate of the transpeptidase reaction |
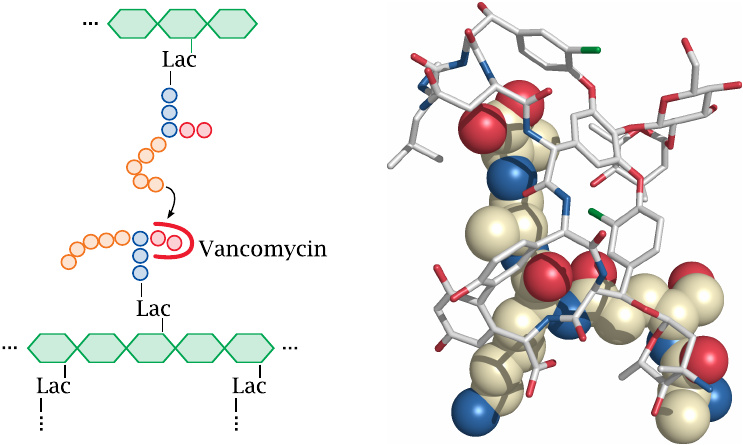
Vancomycin inhibits the same reaction as the β-lactams. However, it does so in a very different and rather unusual manner: Instead of binding to the enzyme, the antibiotic binds to the terminal d-alanine dipeptide on one of the two substrates. Vancomycin is active on Gram-positives but not Gram-negatives.
The structure (rendered from 1gac.pdb) shows not vancomycin itself but the closely related glycopeptide antibiotic A82846B, bound to its substrate. The antibiotic is shown in stick representation, while the peptide moiety of lipid II is shown as spheres. The uppermost part of the peptide consists of the two linked d-alanine residues.
| 11.4.13 |
Vancomycin can be modified to overcome bacterial resistance |
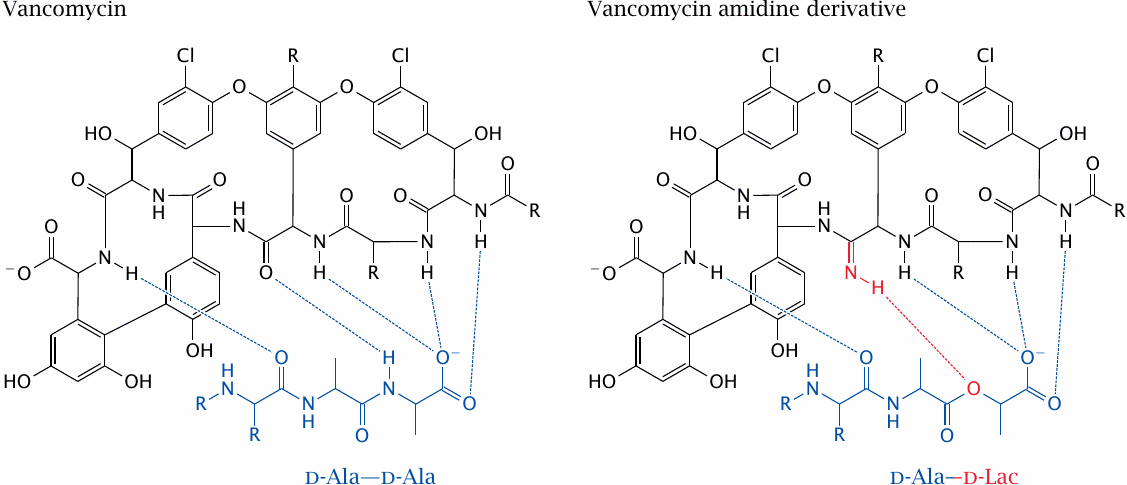
Vancomycin has been widely used in the treatment of serious infections with Gram-positive bacteria, and in particular it has been invaluable in the treatment of MRSA infections. Compared to most other antibiotics, resistance took a long time to develop, which may be related to its unusual mode of action that precludes simple point mutations as a resistance mechanism. However, vancomycin-resistant staphylococci and enterococci have emerged and are spreading. Again through horizontal gene transfer, these resistant bacteria have acquired an enzyme that replaces the terminal d-alanine residue with d-lactate. This removes one hydrogen atom and breaks one of the hydrogen bonds between vancomycin and the substrate, which substantially lowers affinity.
Interestingly, high-affinity binding to the d-alanine–d-lactate substrate can be restored by introducing an amidine group into the vancomycin molecule opposite the ester oxygen in the substrate [97]. Reportedly, the amidine derivative inhibits both vancomycin-sensitive and -resistant bacteria. If a practical and cost-effective method can be found to produce this derivative, it should be of great clinical value.85
Another interesting approach for restoring the activity of vancomycin against resistant bacteria —and one that lends itself more readily to synthesis on a pharmaceutical scale—is the semisynthetic attachment of lipophilic groups to the molecule, whose affinity for the bacterial membrane offsets the decreased affinity for the altered substrate peptide. Several such derivatives are discussed in [98].
| 11.5 |
Antibiotics that inhibit ribosomal protein synthesis |
- Aminoglycosides
- Tetracyclines
- Macrolides
- Chloramphenicol
- Puromycin
- …
Bacterial ribosomes are substantially smaller and structurally different from those found in eukaryotic cells. Many, but not all antibiotics that inhibit ribosomal protein synthesis selectively inhibit the bacterial ones, and can therefore be used for antibacterial chemotherapy in humans. Note, however, that the ribosomes in mitochondria resemble those in bacteria. Inhibition of mitochondrial ribosomes may cause toxicity, and drugs in this class tend to have smaller therapeutic ranges than β-lactams.
| 11.5.1 |
Chloramphenicol lodges into the peptidyl-transfer site of the ribosome |
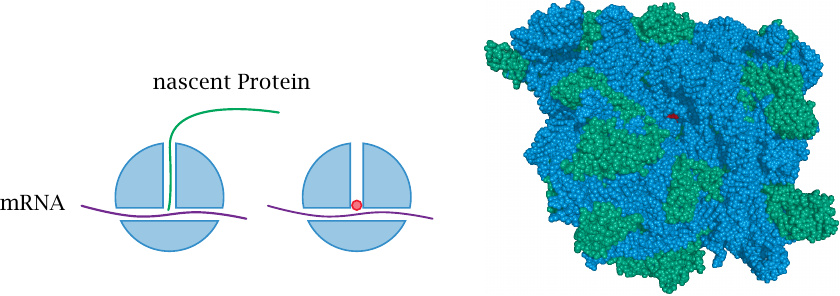
Chloramphenicol binds right within the center of the ribosome and blocks the transfer of the peptide from the peptidyl-tRNA to the next aminoacyl-tRNA. In the ribosome structure on the right-hand side, we are peeping down the peptide exit tunnel to catch a glimpse of the chloramphenicol molecule, which is rendered in red. Ribosomal RNA is shown in blue, and ribosomal proteins are shown in green.
This structure was rendered from 1ko1.pdb. Crystal structures of macromolecules in general are amazing, but the structure of the whole ribosome is a truly mind-boggling accomplishment!
| 11.5.2 |
Structure and action mechanism of puromycin |
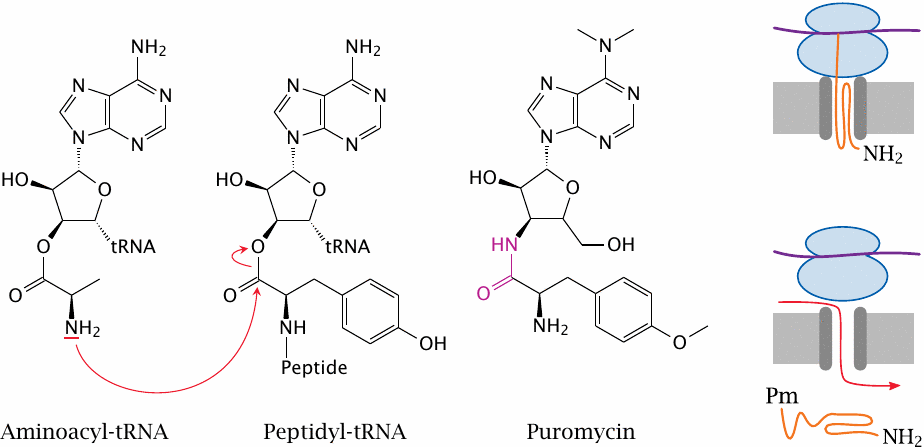
Puromycin disrupts protein synthesis in both prokaryotic and eukaryotic ribosomes, and therefore cannot be used for chemotherapy in humans. However, it has an intriguing structure and mode of action, which has been instrumental in working out the mechanics of protein synthesis itself.
The peptidyl transfer step of protein synthesis involves a single amino acid and the nascent peptide, each of which is bound via an ester bond to a tRNA molecule. In the reaction, the amino group of the incoming amino acid attacks the ester between the growing peptide chain and the corresponding tRNA molecule. The new amino acid residue thereby appends itself to the C-terminus and becomes the anchor of the entire peptide chain. The now empty previous tRNA gets released, and the new peptidyl-tRNA is translocated and awaits attack by the next incoming aminoacyl-tRNA.
Puromycin resembles an aminoacyl-tRNA molecule, and it can therefore attach itself to the C-terminus of a growing peptide chain. However, instead of an ester bond, it contains an amide bond (highlighted) which does not yield to the nucleophilic attack by the next amino acid. Peptide transfer fails, synthesis of the peptide is aborted, and the incomplete peptide is prematurely released.
The disruption of protein synthesis as such is bad enough, but there is more. Proteins that are destined for incorporation into membranes, or for secretion across them, are synthesized at the ER membrane,86 where the ribosome attaches to a so-called translocon pore that guides the nascent peptide into or across the bilayer. Premature peptide release induced by puromycin leaves the translocon in a leaky state and thus renders the membrane permeable towards small solutes.
| 11.6 |
Diverse inhibitors of bacterial macromolecular synthesis |
Ciprofloxacin is a synthetic inhibitor of bacterial DNA topoisomerase, an enzyme which reversibly uncoils the DNA in the tightly packed bacterial chromosome. Inhibition of this enzyme prevents DNA transcription and replication, which is obviously incompatible with life. DNA topoisomerase, or gyrase, inhibitors typically have a broad antibacterial spectrum. Resistance arises through point mutations in the enzyme.87
Rifampicin inhibits RNA polymerase 2, which performs mRNA synthesis. The drug is active against various bacterial species but clinically most valuable in the treatment of mycobacterial infections (tuberculosis and leprosy). Resistance tends to develop rapidly, and combination with other drugs is therefore mandatory.
Fusidic acid inhibits ribosomal protein synthesis by binding to elongation factor 2. It has a sterol ring structure, but bacteria (including Streptomyces species) don’t have sterols, from which one might guess that it is not produced by a bacterium. This is indeed the case; the producer organism is a fungus named Fusidium coccineum.
| 11.7 |
Antibiotics that act on bacterial cell membranes |
Bacterial cell membranes are attractive targets in principle, because they are more exposed and accessible than intracellular targets. However, only few antibiotics that target cell membranes are sufficiently selective for bacterial ones to be of use for antibacterial therapy.
| 11.7.1 |
Structure of the potassium ionophore valinomycin |
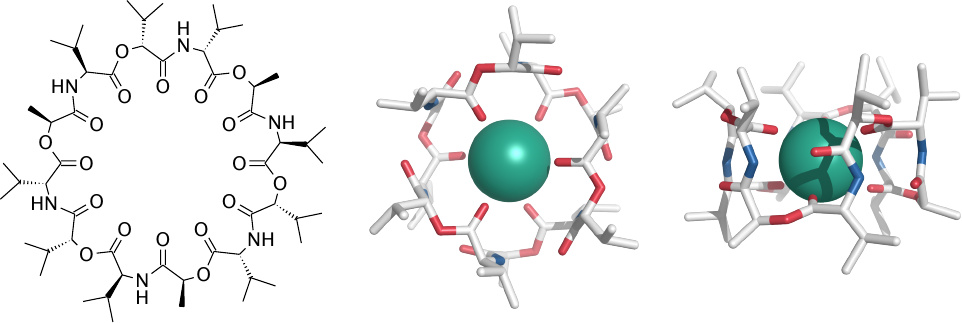
Valinomycin permeabilizes both prokaryotic and eukaryotic cells for potassium and therefore cannot be used for antimicrobial chemotherapy. However, it is useful in biophysical and cell-biological experiments.
Left: The circular molecule contains alternating ester and peptide bonds and exclusively hydrophobic side chains. Right: Valinomycin tightly wraps around a potassium ion, coordinating it with the double-bonded oxygen atoms of the ester bonds. The hydrophobic exterior of the complex enables it the cross cell membranes easily.
| 11.7.2 |
Daptomycin permeabilizes membranes containing phosphatidylglycerol |
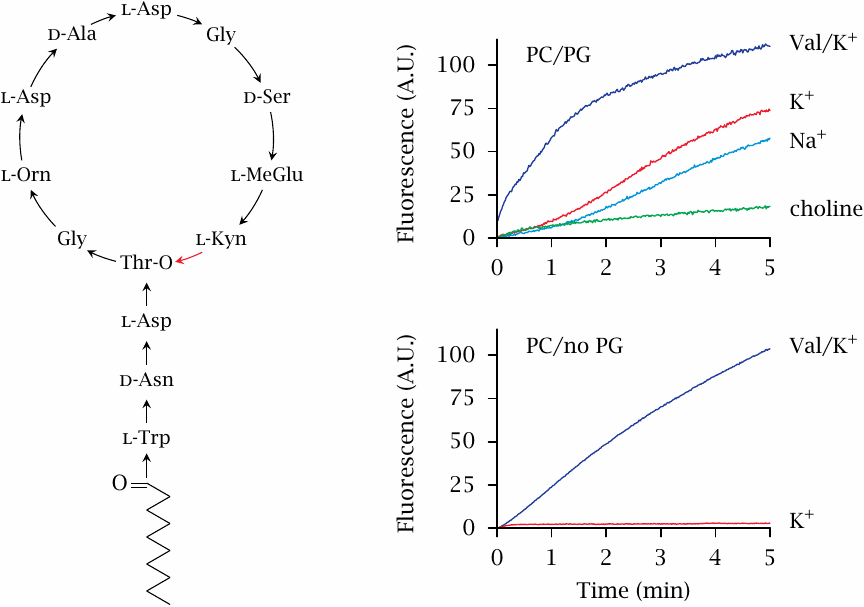
Daptomycin is a lipopeptide antibiotic produced by the soil bacterium Streptomyces roseosporus. It is only active on Gram-positive bacteria, because it fails to penetrate the outer membrane of Gram-negatives. It binds to the cytoplasmic membrane and then forms oligomeric pores [100,101].
The experiment shown here is a fluorescence assay that measures the permeabilization of liposomes by daptomycin. Daptomycin permeabilizes liposomes only when they contain phosphatidylglycerol (PG). This lipid is abundant in the cytoplasmic membranes of bacteria but not in human cell membranes, which is most likely the basis for its selective toxicity. 88 Permeabilization is similarly efficient for K+ and Na+, but considerably less so for choline, which is likely due to the greater size of the choline molecule.
In contrast to daptomycin, valinomycin (Val) permeabilizes both types of membranes, which fits with its known toxicity for both prokaryotic and eukaryotic cells. Figure prepared from original data in [103].
| 11.7.3 |
Structures of polymyxin B and lipopolysaccharide |
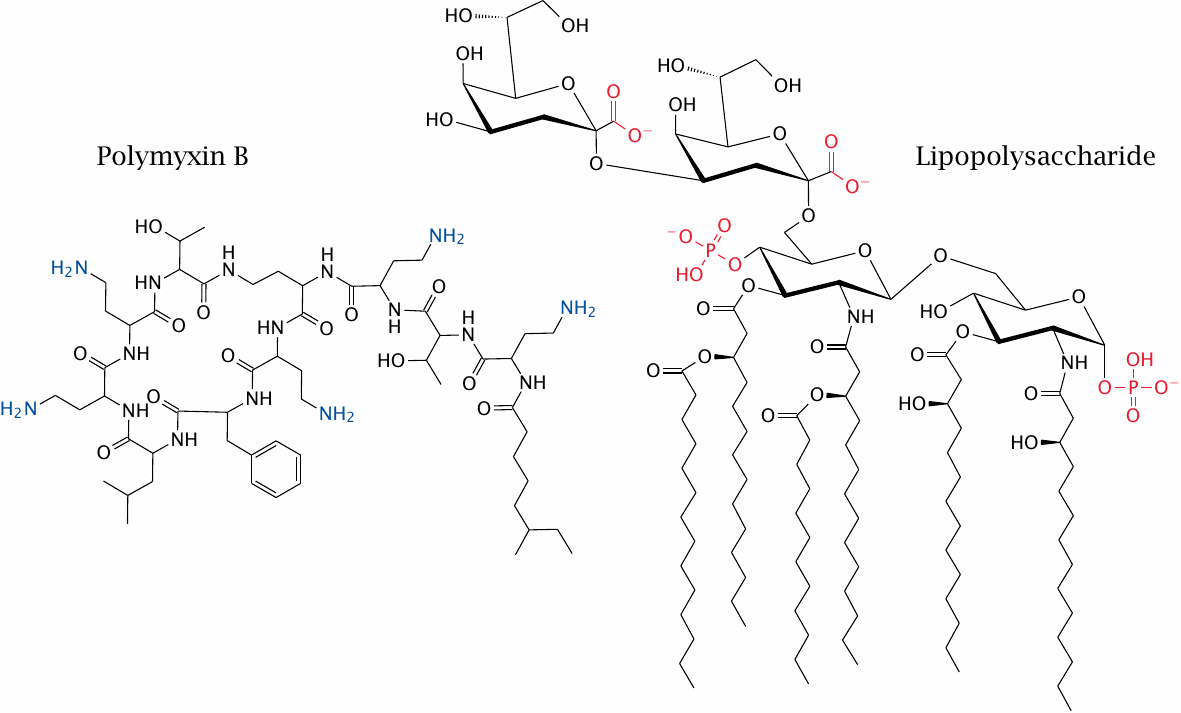
Lipopolysaccharide (LPS) is the major component of the outer leaflet of the outer membrane in Gram-negative bacteria (see slide 11.4.1). The structure shown here depicts the minimal size of the molecule compatible with bacterial life, which has been characterized in biosynthetic mutants; wild type LPS species contain much longer polysaccharide chains that vary in sugar sequence depending on the bacterial species and strain.
Polymyxin B, a lipopeptide antibiotic produced by the Gram-positive bacterium Paenobacillus polymyxa, binds specifically to LPS and disrupts the Gram-negative outer membrane. LPS contains several negative charges, which interact with the positive charges on polymyxin. In addition, there are hydrophobic interactions between the two molecules (see next slide).
| 11.7.4 |
A model of the polymyxin B–LPS complex |
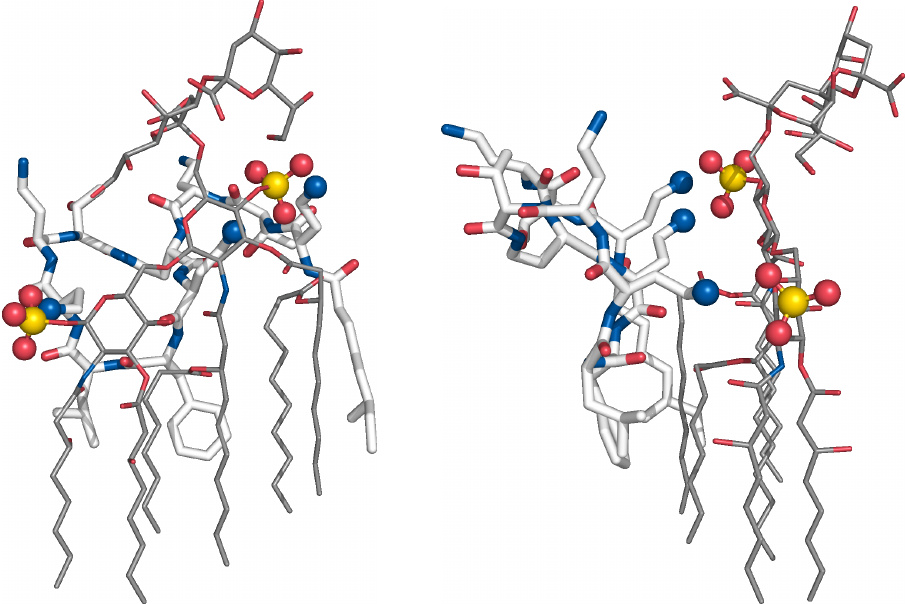
This structure of polymyxin bound to a LPS molecule was not obtained experimentally but through molecular simulation.89
Amino groups in polymyxin B that form ion pairs with the phosphates of lipid A are shown as blue balls. Several hydrophobic side chains of polymyxin B insert between the acyl chains of adjacent lipid A molecules and disrupt the membrane. The lower ends of the acyl chains in LPS, which don’t engage in any contacts with polymyxin, were truncated in this figure.
| 11.8 |
Antifungal chemotherapy |
Drug targets:
- ergosterol in cell membranes (amphotericin B)
- ergosterol synthesis (ketoconazole; terbinafine)
- thymidylate synthase (5-fluorocytosine)
- 1,3-β-glucan synthesis (echinocandins)
Fungi are eukaryotic, and therefore macromolecules like ribosomes and DNA topoisomerase are more similar to those in human cells and not suitable as drug targets for antimicrobial chemotherapy.
Like most bacteria, fungi have cell walls, which contain chitin and 1,3-β-glucan polysaccharides. In addition, fungal cell membranes contain ergosterol instead of cholesterol. The synthesis of cell wall polysaccharide and of ergosterol, as well as the ergosterol-containing bilayer itself, are targeted by antifungal agents.
| 11.8.1 |
Structure and action mode of amphotericin B |
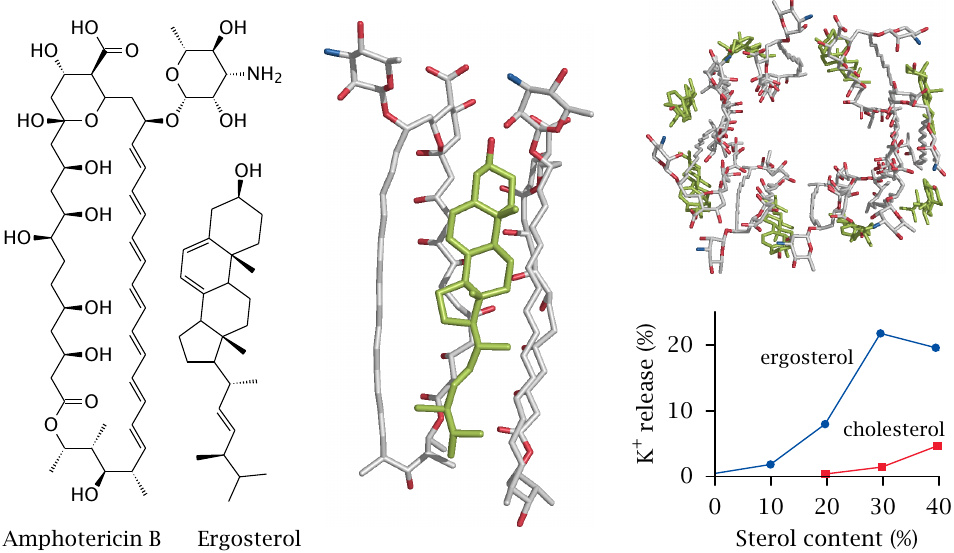
Amphotericin B is a polyene antibiotic produced by a Streptomyces species. It interacts specifically with ergosterol in fungal membranes, and the oligomeric complexes form cation-selective pores in the fungal cell membranes. The top right panel in this slide shows a hypothetical molecular structure of these complexes, as determined by molecular simulation.90 The panel in the middle shows a blow-up from that structure, with one molecule of ergosterol sandwiched between two molecules of amphotericin B.
The plot at the bottom right (redrawn from [104]) shows the release of potassium ions from liposomes varying in sterol content. The extent of release increases with the ergosterol concentration. Liposomes containing high (but physiological) concentrations of cholesterol are also permeabilized.91
The activity of amphotericin B on cholesterol-containing membranes accounts for its toxicity, which can be quite severe. Cell damage is most pronounced in the distal tubules of the kidney, particularly when the pH of the nascent urine is low [105]. Amphotericin toxicity is mitigated when the drug is applied bound to liposomes; presumably, this avoids noxious spikes in the free drug concentration immediately after intravenous application (see slide 14.4.6).
| 11.8.2 |
Other antifungal drugs |
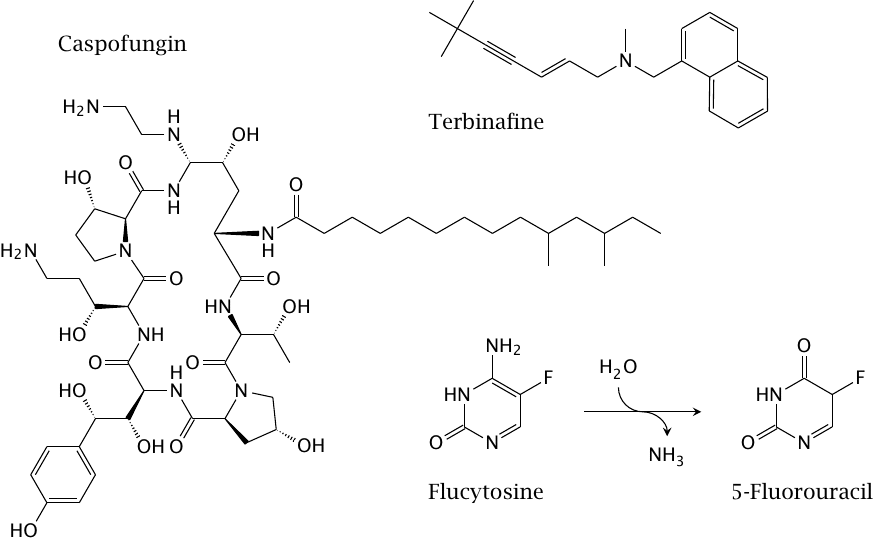
Terbinafine inhibits squalene epoxidase, which catalyzes the cyclization step in the sterol synthesis pathway. This step is common to the synthetic pathways for both cholesterol and ergosterol; therefore, any selective toxicity is not due to absence of this enzyme activity from humans, but rather to molecular differences between the human and the fungal enzymes. Also note that, in contrast to the other drugs listed here, terbinafine is only used externally, so that the question of selective toxicity is not very pressing.
The biosynthesis of ergosterol is also inhibited by ketoconazole and related imidazole derivatives. These drugs inhibit 14α-demethylase, a cytochrome P450 enzyme that catalyzes a step in the conversion of the initial sterol intermediate, lanosterol, to ergosterol. As we had seen earlier, a side effect of ketoconazole is the inhibition of cytochrome P450 enzymes involved in human drug metabolism (see slide 4.2.4); therefore, the modes of action and of toxicity are related.
Flucytosine is a prodrug that in fungal cells undergoes deamination to fluorouracil, which inhibits thymidylate synthase (see slide 12.6.2). Human cells do not deaminate the flucytosine, which is the basis for selective toxicity. Note, however, that the deaminase enzyme is not essential for fungal cells either, and mutational inactivation of the enzyme enables rapid development of resistance. Therefore, flucytosine is only useful in combination with other antifungal drugs.
Caspofungin is a lipopeptide antibiotic of the echinocandin type that inhibits an enzyme involved in the biosynthesis of 1,3-β-glucans, an essential component of the fungal cell wall.
| 11.9 |
Antiprotozoal drugs |
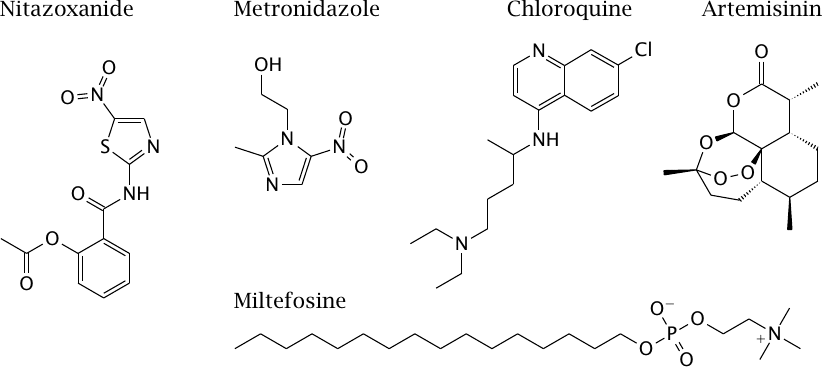
As noted earlier, the term “protozoa” covers a pretty diverse lot of single-celled pathogens, and so it is not surprising that there is no such thing as a broad-spectrum antiprotozoal drug. We will here look at a few example drugs with different and interesting modes of action.
The plant alkaloid artemisinin inhibits a sarco-endoplasmatic Ca++-ATPase (SERCA) in malaria parasites; this pump is essential for transporting Ca++ from the cytosol into the ER and thereby terminating calcium signals. Its recently increasing use has already given rise to resistance that has been traced to point mutations in SERCA [106]; this association has been disputed, however [107], which suggests the existence of alternate action mechanisms.
Miltefosine is a lipid analogue that inhibits cytochrome C oxidase [108]. Metronidazole and nitazoxanide are reduced to toxic nitro radicals by anaerobic metabolism; this is explained below.
| 11.9.1 |
Chloroquine inhibits formation of β-hematin |
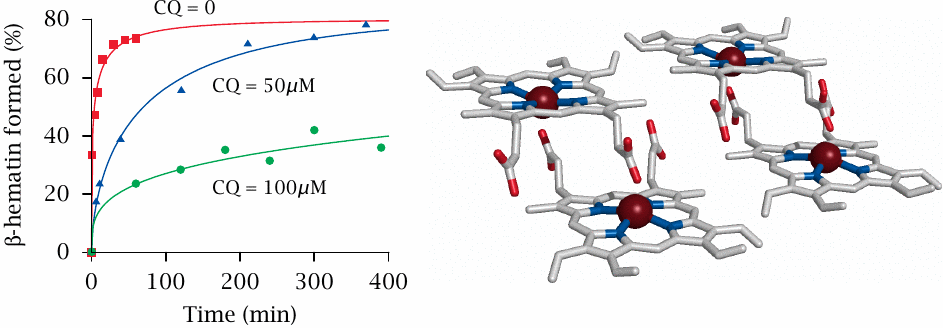
Chloroquine was developed in the 1930s and came to be widely used during World War II. For several decades, chloroquine remained the standard drug for prevention and treatment of malaria. Its mechanism of action is quite unusual and indirect.
Malaria plasmodia that live and grow in erythrocytes feed on hemoglobin, or rather only its protein component (globin). Free heme is left over and is toxic to the parasites, because it catalyzes the formation of reactive oxygen species from molecular oxygen.92 To combat heme toxicity, malaria parasites induce its crystallization (right); these β-hematin or hemozoin crystals are visible in blood smears as the so-called malaria pigment.93 Chloroquine interacts with the surfaces of growing β-hematin crystals, inhibits deposition of further heme molecules (left), and it thereby indirectly promotes formation of toxic reactive oxygen species.94
The accumulation of chloroquine inside the parasite’s vacuole is driven by a pH gradient—the interior of the vacuole is acidic, which retains the basic drug molecule by way of non-ionic diffusion (see slide 3.4.7).
Considering the immutable nature of its target, it is clear that resistance to chloroquine must be mediated by removal or inactivation of the drug. Indeed, the most important mechanism of resistance consists in extrusion from the plasmodial cells by ABC type transporters. Resistance is now widespread among the parasites.
| 11.9.2 |
Action mechanism and selective toxicity of nitroimidazoles |
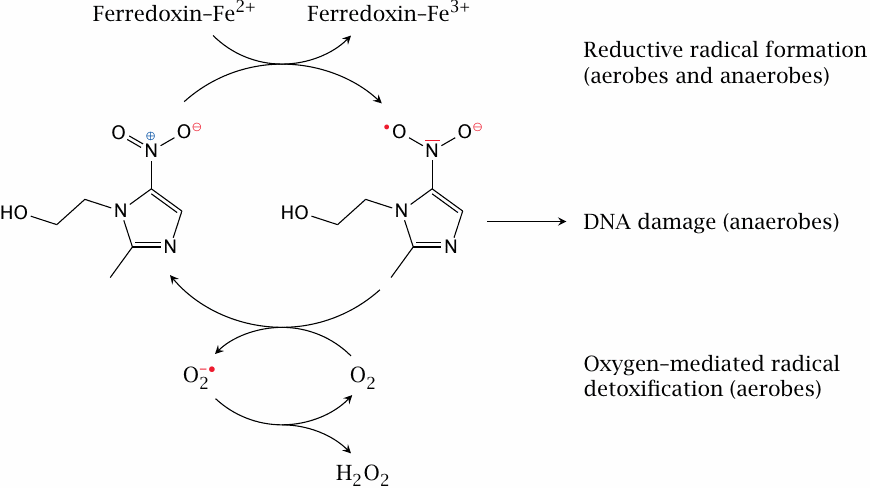
Nitroimidazoles are active on protozoa whose energy metabolism is anaerobic and which contain hydrogenosomes. These organelles produce ATP from the conversion of pyruvate to acetate, CO2, and H2. Hydrogenosomes contain high activities of the redox-active protein ferredoxin [112]. Reduction of nitroimidazoles by ferredoxin produces nitro radicals which then attack the parasites’ DNA; intriguingly, the extent of damage to the DNA is strongly correlated with A/T content [113]. Interestingly, nitroimidazoles are also active and clinically used against anaerobic bacteria such as Bacteroides fragilis.
Mammalian cells don’t produce large amounts of nitroimidazole radicals, and in addition they are able to detoxify them through reaction with molecular oxygen. The superoxide formed in the latter reaction is then scavenged by superoxide dismutase.
| 11.9.3 |
Melarsene oxide binds trypanothione |
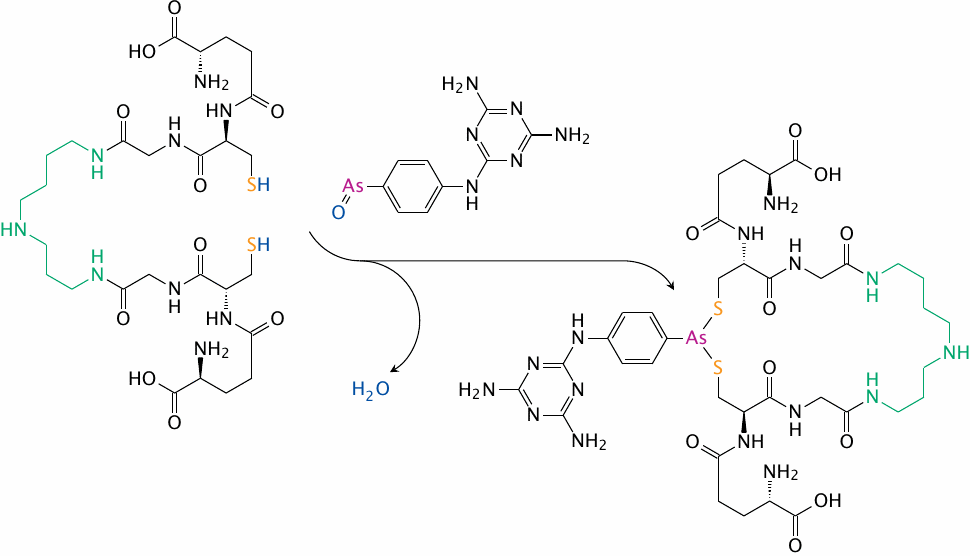
Organic arsenic compounds such as melarsene oxide are used in the treatment of infections caused by Trypanosoma species that are transmitted by tsetse flies and cause sleeping sickness.
The major intracellular thiol in Trypanosoma cells is trypanothione, which consists of two molecules of glutathione that are connected by a spermidine linker, which is shown here in green. Trypanothione and melarsene oxide form a chelate [114] , which then acts as an antimetabolite to the free thiol itself in trypanothione-dependent reductive enzyme reactions. This causes oxidative damage to the cell. Resistance of the parasites to melarsene oxide is due to increased production of trypanothione and to active extrusion of the complex [115].
| 11.10 |
Antiviral chemotherapy |
Viruses are heterogeneous in more than one way. They differ greatly in size; small viruses measure less than 30 nm across and contain only a handful of genes, whereas large ones approach bacteria in size and genomic complexity. The genome may be single- or double-stranded, consist of RNA, of DNA, or of both. It may be packaged as a single molecule or in multiple segments; if the former, the viral proteins may be translated separately or in the form of single precursor polyprotein. The virus particle may be coated with a protein capsid only, or the capsid may be surrounded by a lipid membrane. Finally, replication may occur entirely the cytoplasm or in part in the nucleus.
Like the viruses themselves, strategies for chemotherapy are diverse. Here, we will not consider them systematically but only by example.
| 11.10.1 |
The life cycle of influenza virus |
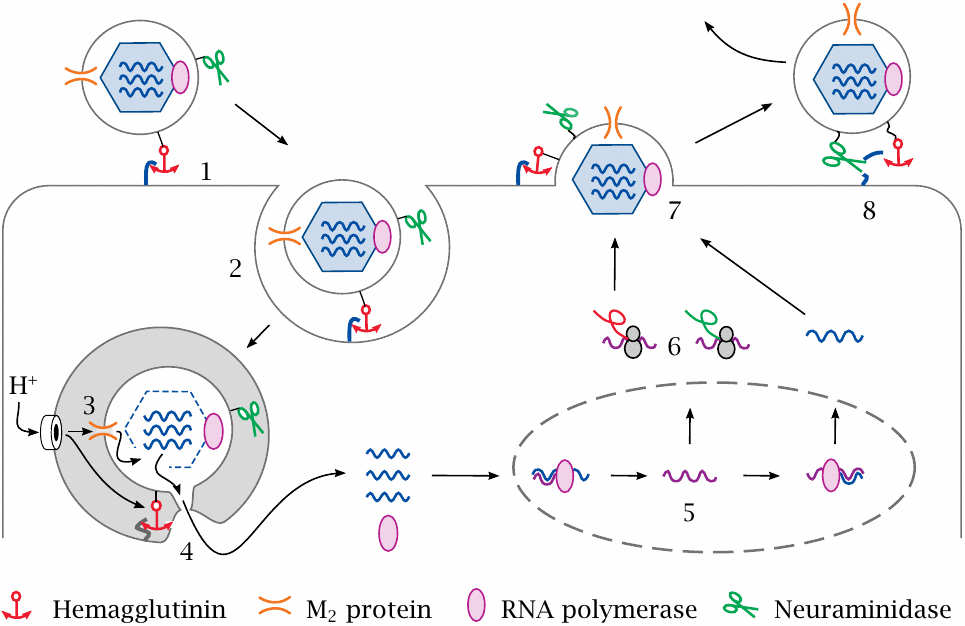
Influenza virus contains a segmented minus-strand RNA genome and is enveloped by a lipid membrane. Embedded in the lipid membrane are several proteins that fulfill their respective functions in different stages of the viral life cycle.
In the first stage of infection, the virion attaches to neuraminic acid residues on the target cell via its hemagglutinin surface protein (1). It is then taken up by endocytosis (2), and the cell begins to pump protons into the endocytotic vesicle. Protons enter the virus particle through the M2 channel (3), which induces the viral protein capsid to disintegrate and release the viral RNAs.
Acidification of the vesicle also triggers the membrane fusion activity of the viral hemagglutinin (4), which creates a passageway through which the uncoated viral RNAs and the RNA polymerase enter the cytosol. Both translocate to the nucleus (5), where the viral RNA is transcribed and replicated. Plus strand RNA transcripts are translated by ribosomes in the cytosol (6). Progeny virions assemble and bud at the cytoplasmic membrane (7), from which they are released by neuraminidase (8).
The drugs shown in the next two slides interfere with step (3) and step (8) of this life cycle, respectively. Nucleoside analogues that inhibit the RNA polymerase (step 5) are in clinical use as well but are not shown here.
| 11.10.2 |
Amantadine blocks the M2 proton channel |
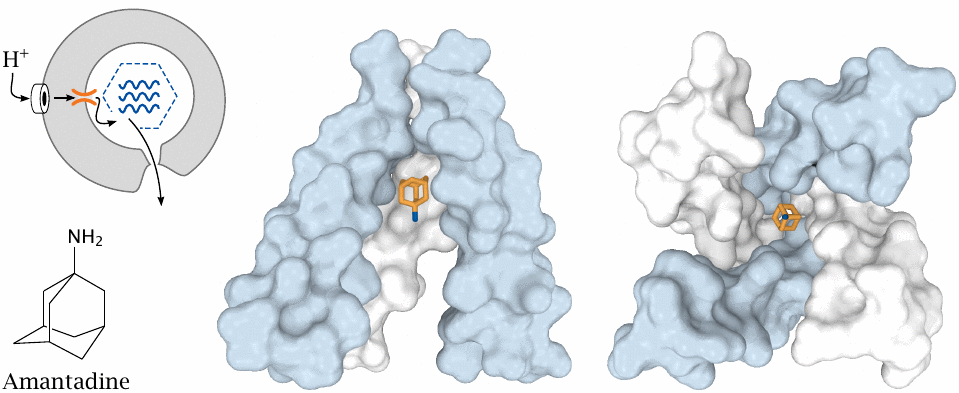
As we have just seen, uncoating and release of the viral RNA requires that protons reach the interior of the virion through the M2 proton channel. The drugs amantadine and rimantadine block this channel and therefore disrupt this step of the viral life cycle.
Center: Side view of amantadine bound in the cavity of the channel. One of the four subunits has been removed to provide a view into the channel interior. Right: View into the blocked channel cavity from inside the virus particle. Structures rendered from 3c9j.pdb.
| 11.10.3 |
Oseltamivir inhibits influenzavirus neuraminidase |
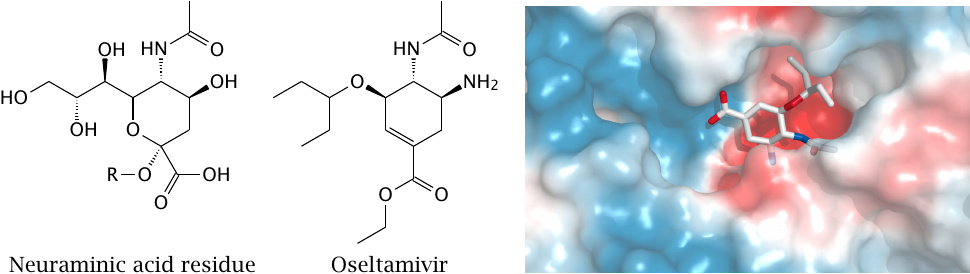
Neuraminidase is important in the final step of viral life cycle, that is, in detaching the particle from the expiring host cell. The enzyme cleaves neuraminic acid from the free end of a cell surface oligosaccharide, which is represented by R in the formula of neuraminic acid.
Oseltamivir mimics the transition state of neuraminic acid that occurs in the cleavage reaction. The drug is shown here bound within the active site of neuraminidase (rendered from 2hu4.pdb). To the left of the cavity occupied by oseltamivir, another, unoccupied groove is visible that presumably accommodates the adjacent sugar moiety of the substrate polysaccharide.
| 11.10.4 |
Inhibition of HIV fusion with target cells by the peptide enfuvirtide |
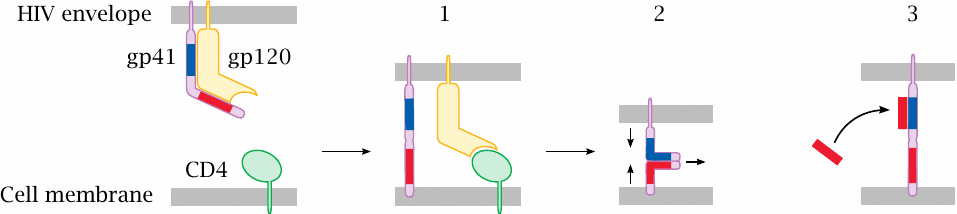
While HIV (human immunodeficiency virus) is not related to influenza virus, it also contains a lipid membrane, which must fuse with the host cell membrane to initiate the infection process. Fusion is controlled by two HIV surface glycoproteins, gp41 and gp120, which initially are associated with each other. Binding of gp120 to its cellular receptor, CD4, and to a co-receptor (not shown) releases the fusion protein gp41, the tip of which inserts into the target cell membrane (1). Two complementary heptad repeat motifs in gp41 (red and blue) then zip up against one another, causing the two membranes to converge (2) and eventually fuse. The drug enfuvirtide, a synthetic peptide, resembles one of these repeats and accordingly associates with the other, which disrupts gp41 activity (3).
While—ostensibly for the sake of simplicity, but really due to a limited supply of artistic talent—gp41 was rendered here as a monomer, it is actually a trimer. Accordingly, polymeric analogues of enfuvirtide have shown superior affinity and inhibition of cell infection [116].
| 11.10.5 |
Inhibitors of virus genome replication |
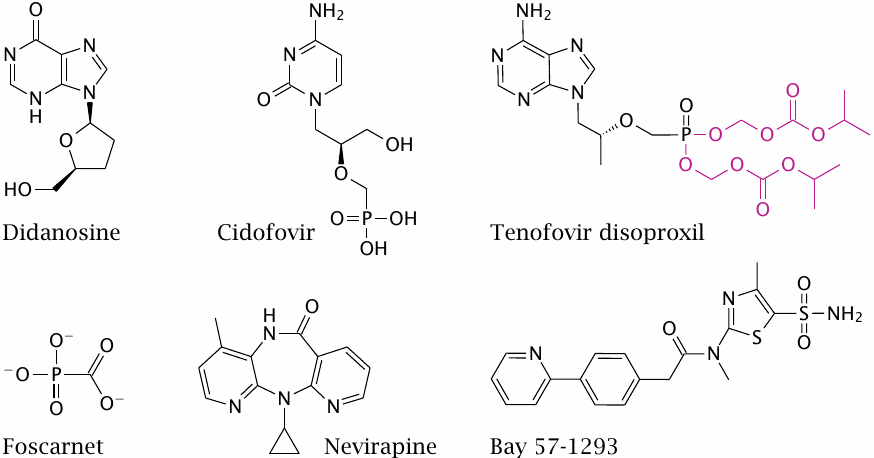
The most general approach to antiviral chemotherapy remains the inhibition of the viral DNA or RNA polymerases that replicate the viral genomes.
Didanosine and tenofovir disoproxil are chain terminators, and nevirapine is an allosteric inhibitor of HIV reverse transcriptase. Didanosine is phosphorylated by cellular kinases.95 Tenofovir disoproxil is a resorption ester of a phosphonate analogue of adenosine. Cidofovir, which is active against many viruses, likewise carries a phosphonate group and therefore bypasses the first phosphorylation step.
While most inhibitors resemble substrates of nucleic acid synthesis, foscarnet resembles a product. Pyrophosphate is produced in each addition of a nucleotide to a growing nucleic acid strand and then rapidly hydrolyzed by pyrophosphatase. On several viral polymerases, the hydrolysis-resistant analogue foscarnet blocks the binding site for pyrophosphate, at concentrations below those that would be inhibitory for human polymerases.
Bay 57-1293 is an experimental inhibitor of Herpesvirus helicase/primase, an enzyme that unwinds DNA and synthesizes a short oligonucleotide primer for the DNA polymerase.
| 11.10.6 |
Activation of acyclovir |
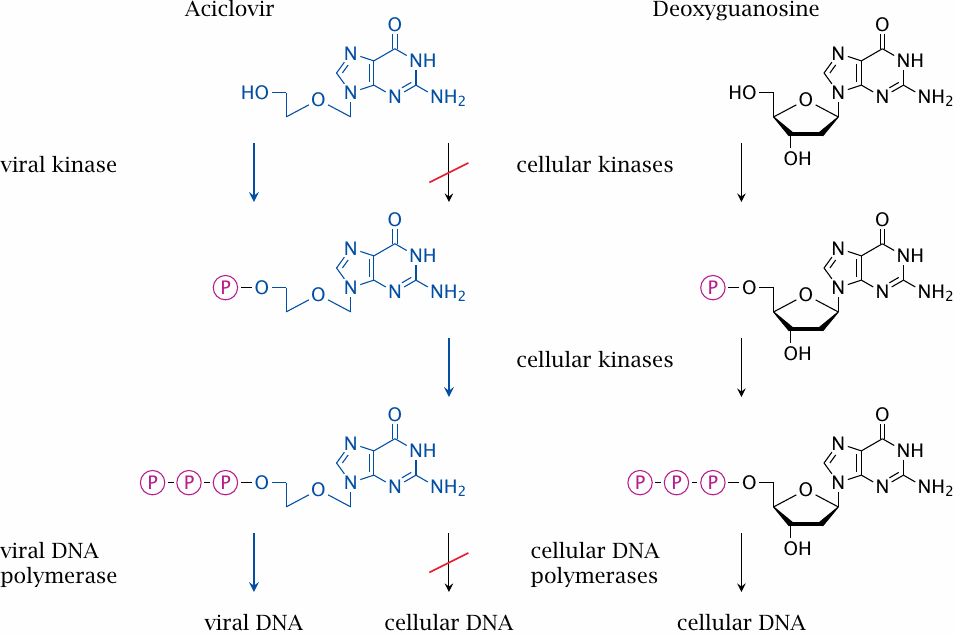
Acyclovir is a guanosine analogue that has been mutilated to a considerable extent, and accordingly is rejected as a substrate by cellular nucleoside salvage kinases. However, it is phosphorylated by a thymidine kinase encoded by herpes virus. Cellular nucleotide kinases then convert the monophosphate to the triphosphate, which is a substrate for the viral DNA polymerase, but not for the cellular enzyme.
In contrast to most antimicrobial drugs, which bind to a single microbial target, acyclovir selectively interacts with two viral enzymes in series. Because of this, acyclovir has a particularly high degree of selective toxicity, and serious side effects are less common with acyclovir than with most other nucleoside analogues used in antiviral chemotherapy.
| 11.10.7 |
Function of virus proteases |
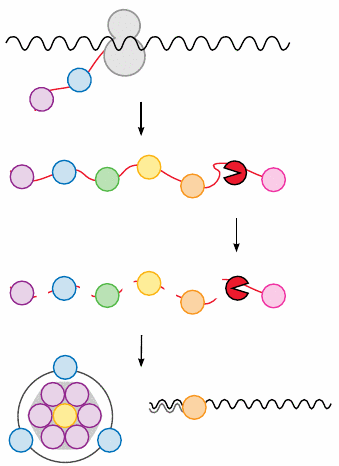
Many viruses, including hepatitis C virus and HIV, translate their genome into polyproteins, that is, large precursor proteins which contain the final, functional proteins as sub-domains like pearls on a string.
One of these sub-domains is a protease that cleaves first itself and then the other components. After cleavage, the released proteins travel to different destinations to serve in their respective roles during virus replication and assembly.
If the viral protease inhibited, most viral proteins will not become functional; in particular, assembly of the virus particle will be prevented. Inhibition of this crucial step is therefore extremely disruptive to viral replication.
In the case of HIV, the advent of protease inhibitors marked a very major improvement in the effectiveness of treatment, causing the life expectancy of the patients to jump up from just a few years after diagnosis to an almost normal life span. The HIV protease inhibitor saquinavir is shown in slide 1.3.6.