| 4 |
Drug metabolism |
| 4.1 |
Overview |
In the last chapter, we considered the renal elimination of drugs. While some drugs are eliminated via the kidneys unchanged, most of them undergo metabolic transformation at least in part before being excreted.
| 4.1.1 |
Types of reactions in drug metabolism |
- Oxidation
- Conjugation
- Reduction
- Hydrolysis
These categories of metabolic transformations are very broad; for each category, there is a multitude of enzymes and specific reactions. We have already seen examples of drug metabolism by hydrolysis, which is exploited in the design of resorption esters (see slide 3.4.5). As an example of reductive metabolism, we have seen the activation of sulfamidochrysoidine via azoreduction (see slide 1.3.3). As we will see in this chapter, oxidative metabolism and conjugation reactions are even more common.
| 4.1.2 |
Functional outcomes of drug metabolism |
- Inactivation and accelerated elimination of drugs
- Activation of prodrugs
- Formation of active metabolites with similar or novel activity
- Detoxification of toxic xenobiotics
- Toxification of non-toxic xenobiotics
Drugs are a subset of xenobiotics, that is, exogenous substances. Examples of xenobiotics other than drugs are plant alkaloids or fungal toxins. The metabolic pathways and enzymes that have evolved to dispose of these natural xenobiotics have broad substrate specificity, and they are therefore active on many synthetic drugs also.
In most cases, drug metabolism results in inactivation and accelerated elimination of drugs from the body. However, as this list indicates, other outcomes of metabolism are possible; we will see examples below.
In the list above, outcomes 2. and 3. both yield active products. The difference between them is that, in 3., both the original drug and the metabolite have pharmacological activity. In contrast, a prodrug has no activity before undergoing metabolic conversion.
| 4.1.3 |
A hydrolytic metabolite of cocaine can be detected in wastewater |
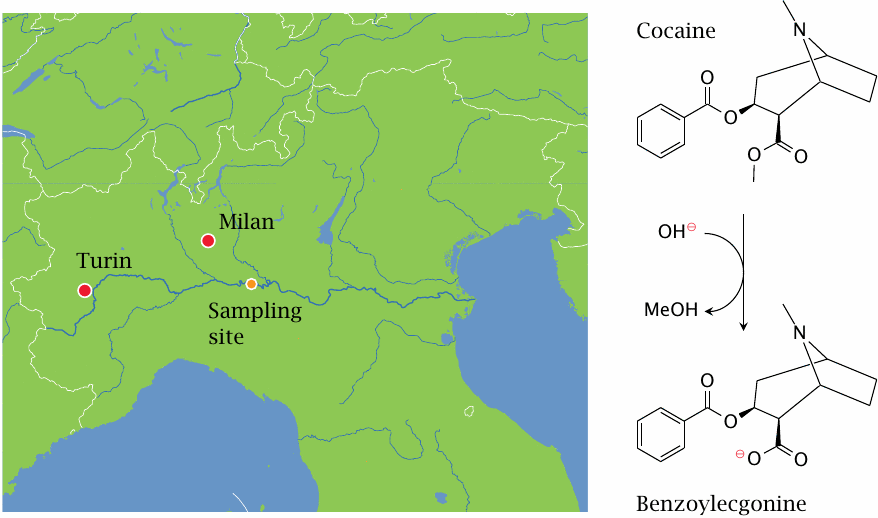
Cocaine is inactivated by hydrolysis. The metabolite, benzoylecgonine, is excreted with the urine. It is fairly stable and thus can be detected in wastewater.
A study from Italy [18] analyzed water samples from the river Po, which (together with the flow rate of the river) were used to estimate the consumption of cocaine upstream of the sampling site. The samples were taken at a point downstream of Turin and, in part, of Milan, two of Italy’s four largest cities. Cocaine consumption in this area was found to be 4–5 times higher than had previously been estimated. Similar findings have since been reported from other European countries.
| 4.1.4 |
Metabolism of phenobarbital |
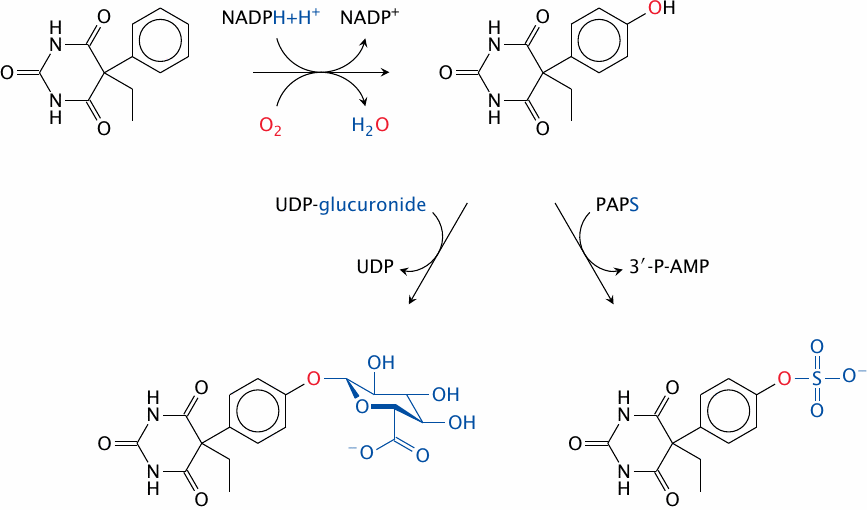
Phenobarbital is a barbituric acid derivative. It is an agonist at the GABAA receptor (see slide 6.10.8) that is sleep-inducing and antiepileptic. The drug molecule itself is quite hydrophobic; this enables it to penetrate the blood brain barrier, and it also slows down renal elimination. Therefore, 75% of the compound is excreted in the form of conjugated metabolites.
Phenobarbital as such, however, is not a good substrate for conjugation reactions. To make it amenable to conjugation, a suitable functional group—an aromatic hydroxyl group—is first introduced by a cytochrome P450 enzyme. Aromatic or aliphatic hydroxylations by such enzymes are very common phase I reactions.
In phase II of phenobarbital metabolism, the hydroxyl group just introduced is conjugated either with glucuronic acid by a UDP-glucuronosyltransferase, or with sulfate by a sulfotransferase. Both of these metabolites are fairly polar and are effectively excreted through the kidneys.
| 4.1.5 |
With many drugs, metabolic transformation facilitates excretion |
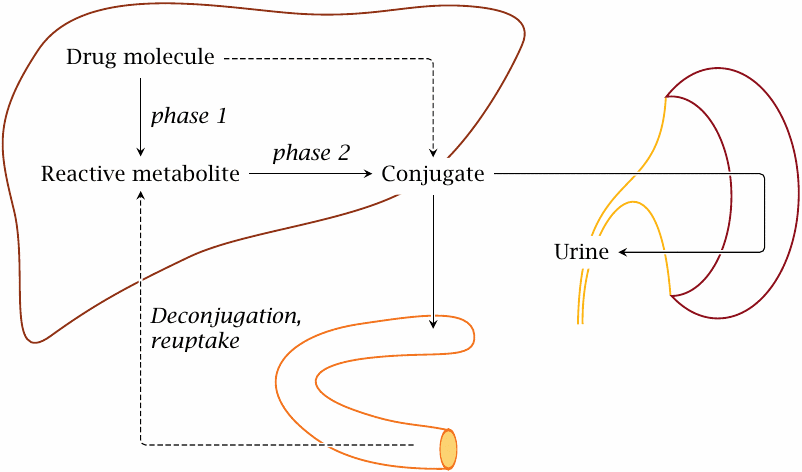
The metabolism of phenobarbital illustrates a pattern that applies to many other drugs as well. With many drugs, hydroxylation by cytochrome P450 or some other phase I reaction precedes conjugation with glucuronic acid, glutathione, or another functional group. These conjugations are collectively referred to as phase II reactions. Excretion of conjugated drugs is sometimes referred to as phase III of drug elimination.
The most important organ in drug biotransformation is the liver. The conjugated drugs may be exported into the blood stream and be eliminated in the kidneys, or they may be excreted into the bile and then reach the intestine. In the latter case, they may undergo deconjugation by bacterial enzymes in the large intestine and subsequent reuptake. Reabsorbed drugs will be transported to the liver via the portal vein and may undergo another round of conjugation and biliary excretion; this is called entero-hepatic cycling.
| 4.1.6 |
Hydrophobicity does not strongly predict the extent of metabolism |
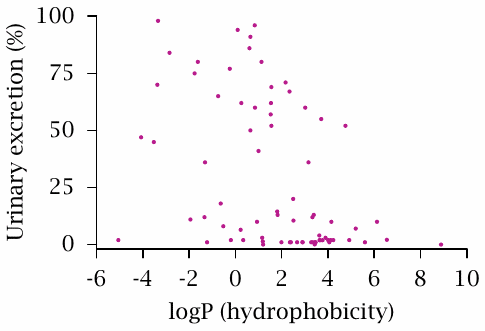
This scatter plot of a randomly selected sample of 75 drugs (data from [19]) correlates the percentage of a drug that appears in the urine in unchanged form to the estimated logP value. The logP values is the logarithm of the octanol/water partitioning coefficient; therefore, drugs with high logP values are hydrophobic.
There may be a very slight trend for hydrophobic drugs to undergo renal elimination to lesser extent; however, regardless of logP, only very few drugs are quantitatively eliminated through the kidneys. In most cases, the remainder will undergo metabolic transformation, although a few drugs may be excreted into the bile unchanged.
So, the key point of this slide is not in the correlation of the two parameters, but rather in the fact that most drugs undergo metabolic transformation at least in part; and contrary to lore, this applies to both hydrophilic and hydrophobic drugs.
| 4.1.7 |
Major types of drug-metabolizing enzymes |
Phase I
- Cytochrome P450 enzymes
- Diaphorase (NADH:quinone oxidoreductase)
Phase II
- UDP-glucuronosyltransferases
- Sulfotransferases
- Glutathione-S-transferases
- N- and O-acetyltransferases
Cytochrome P450 enzymes are the most important class of enzymes in phase I metabolism. Other oxidative enzymes in drug metabolism include flavin monooxygenase and monoamine oxidase. Reductive metabolism is carried out by various enzymes; diaphorase, also called quinone reductase, is a major one.
In addition to glucuronosyl- and sulfotransferases, glutathione-S-transferases and acetyltransferases also contribute significantly to phase II metabolism. Methyl conjugation and amino acid conjugation are less common.
While the number of enzymes involved in the degradation of xenobiotics is large, it is necessarily far smaller than the virtually limitless number of potential substrates. Therefore, many of these enzymes must be able to modify a large number of structurally diverse drugs and xenobiotics. The same also applies to transporters involved in phase III, that is, biliary and urinary excretion.
| 4.2 |
Cytochrome P450 enzymes |
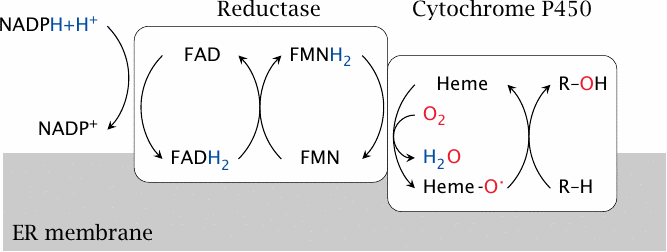
Cytochrome P450 isoforms are associated with the membrane of the endoplasmic reticulum (mostly) or the mitochondria (some). Membrane association facilitates interfacial processing of lipophilic substrates that often reside in the apolar phase of the membrane.
Cytochrome P450 enzymes cooperate with cytochrome P450 reductase, which acquires two electrons from NADPH and, via two flavin coenzymes,25 passes them on one by one to the heme group in the cytochrome. Thereafter, they are used to reduce one of the oxygen atoms of O2 to water. The second oxygen atom is retained in a highly reactive form, and subsequently carries out the actual attack on the substrate.
| 4.2.1 |
Reactions catalyzed by cytochrome P450 |
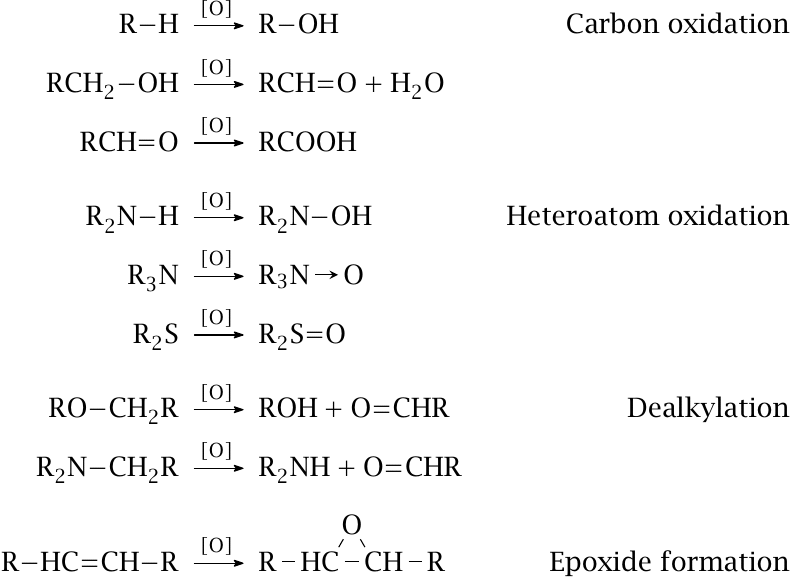
Halfway through the reaction, the active site of cytochrome P450 contains a single oxygen atom in a loosely bound, highly reactive state. This oxygen atom can react with many different functional groups, and in various ways, as summarized on this slide. The particular reaction that occurs with a given substrate depends more on which of its functional groups happens to be the nearest to the oxygen, rather than on which one might be the most reactive itself.
The next slide presents some examples of drugs that undergo one or the other of the cytochrome P450-mediated oxidative reactions shown here. In those examples, the reaction products also happen to be active metabolites; this, however, is not always the case.
| 4.2.2 |
Formation of active metabolites by CYP450 enzymes |
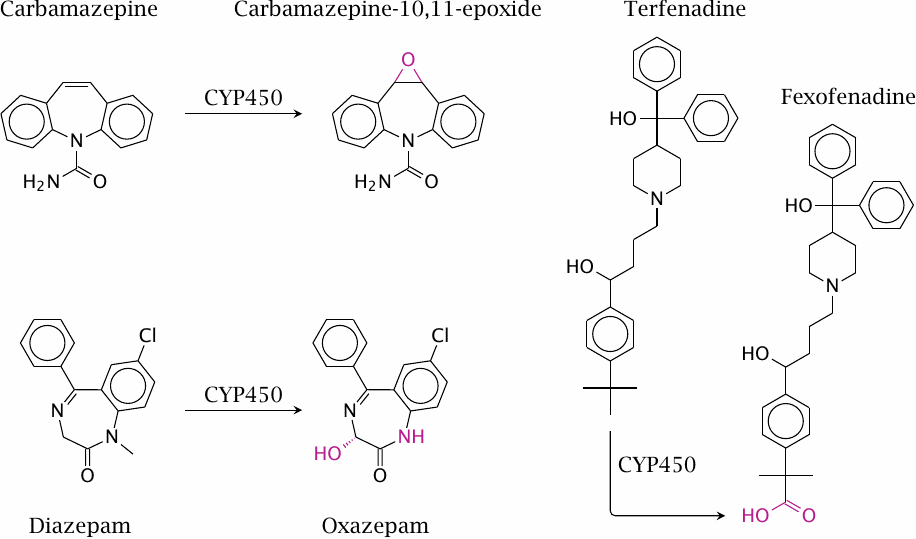
Diazepam is an agonist of the GABAA receptor in the brain (see slide 6.10.8) that is used as a tranquilizer and in some forms of epilepsy. Both the hydroxylation and the demethylation of diazepam are performed by cytochrome P450 enzymes. The resulting metabolite, oxazepam, is also used as a drug in its own right. Since it no longer has to undergo phase I metabolism, it is conjugated and eliminated more rapidly than is diazepam, which is advantageous for example when used as a sleeping aid.
Carbamazepine blocks sodium channels (slide 6.5.4); this is useful in epilepsy and several other neurological conditions. The epoxide metabolite of carbamazepine significantly contributes to the pharmacological activity, but also to the toxicity, of carbamazepine. Like other epoxides, carbamazepine-10,11-epoxide may wreak havoc by reacting covalently with intracellular nucleophiles.
Fexofenadine and terfenadine are histamine H1 receptor blockers. Fexofenadine was originally observed as an active metabolite of terfenadine. The metabolite has since supplanted the original drug, because it avoids some toxic side effects on cardiac excitation that may occur when the parent compound accumulates due to inhibition of cytochrome P450-mediated metabolism. Such inhibition can arise from drugs such as ketoconazole (see below) but also from regular foods such as grapefruit [20].
The metabolism of terfenadine also illustrates that it is possible for one substrate molecule to undergo several rounds of oxidation—in this case, all the way from a methyl to a carboxyl group—that will most likely occur in one sitting.
| 4.2.3 |
Erythromycin bound to the active site of cytochrome P450 |
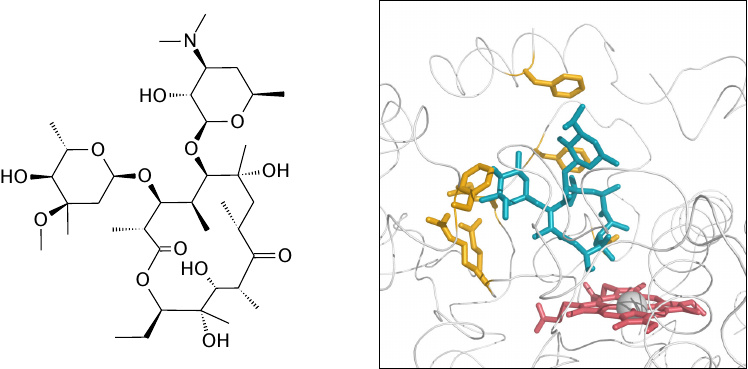
This slide and the following ones illustrate the promiscuous substrate binding by cytochrome P450, subtype 3A4. The enzyme was crystallized in the presence of erythromycin or of ketoconazole, respectively. Erythromycin is shown in this figure, ketoconazole in the next one. Erythromycin is shown in blue, heme in red, and amino acid side chains in close contact with the drug are shown in yellow. Figures rendered from 2j0d.pdb and 2v0m.pdb [21].
| 4.2.4 |
Ketoconazole bound to the active site of cytochrome P450 |
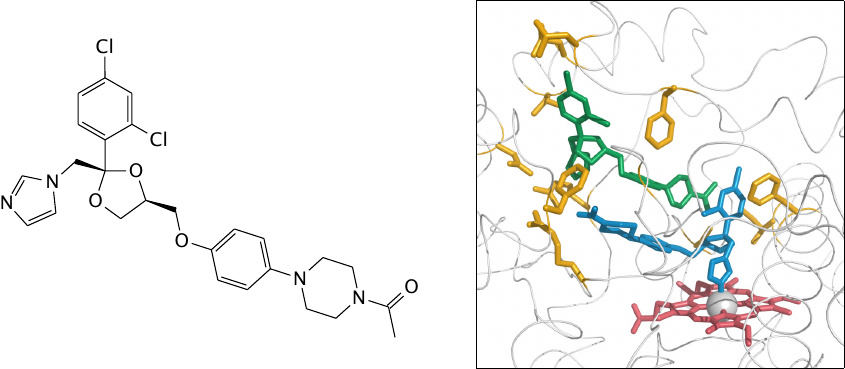
In this structure, two molecules of the antifungal drug ketoconazole (shown in blue and green) are bound in the active site; the orientation of the upper one is similar to that of the structural formula.
The binding of two drug molecules to one enzyme molecule is likely caused by the high concentrations used in the crystallography experiment; in vivo, only one molecule would likely be bound. Nevertheless, this structure does illustrate the enzyme’s ability to accommodate substrates with rather diverse structures.
Note the participation of different side chains in binding erythromycin and ketoconazole, and also the direct association of the ketoconazole molecule shown in blue with the catalytic heme. Ketoconazole is not only a substrate but also an inhibitor of cytochrome P450; this inhibitory action can cause side effects in humans but is also responsible for the antifungal activity of the drug (see Section 11.8.2).
| 4.2.5 |
Superposition of the erythromycin- and the ketoconazole-bound CYP3A4 structures |
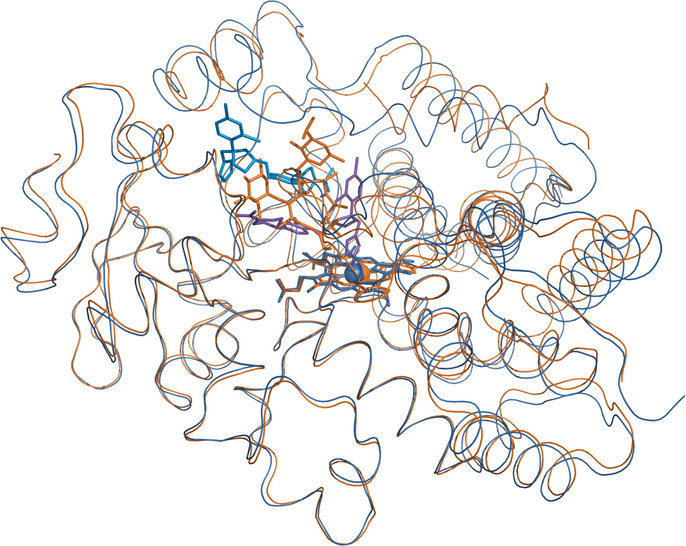
The erythromycin-bound structure is shown in orange, the ketoconazole-associated one in blue. The polypeptide backbones track each other for the most part but diverge noticeably in several places, particularly atop the active site. These local deviations illustrate the conformational flexibility of the enzyme that allows it to accommodate many different substrates.
Cytochrome P450 3A4 (CYP3A4), the subtype shown in these two structures, is involved in the metabolism of as many as 50% of all clinically applied drugs. While this observation also suggests an astonishingly broad substrate specificity, another reason for the prominent role of CYP3A4 in drug metabolism is its upregulation in the presence of drugs, which is mediated by transcriptional induction.
| 4.3 |
Transcriptional induction of drug metabolism |
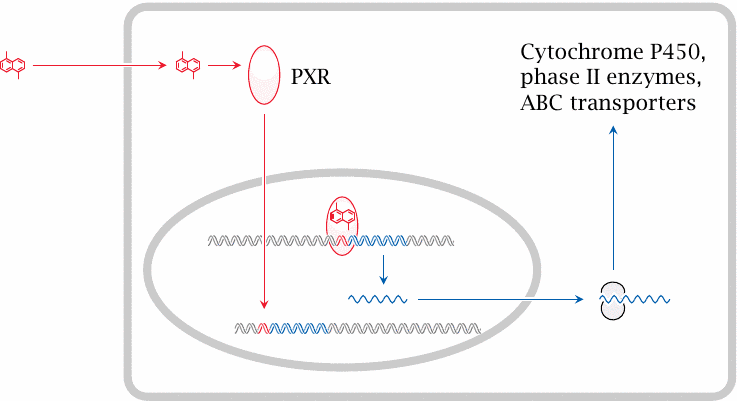
The transcriptional induction of CYP3A4 and several other drug-metabolizing enzymes and drug-excreting transporters is mediated by the pregnane X receptor (PXR). This receptor belongs to the family of nuclear hormone receptors, which also comprises the receptors for thyroid and steroid hormones (see chapter 7). The PXR binds to a wide range of drugs. Upon drug binding, the receptor translocates from the cytosol to the nucleus and binds to its cognate regulatory DNA sequences, named xenobiotic response elements (XRE), and thereby causes increased transcription of genes in the vicinity.
Induction of drug-metabolizing enzymes often leads to accelerated metabolism of multiple drugs, not just the inducing drug itself. For example, rifampicin, an antibiotic used in tuberculosis, or phenytoin and phenobarbital, which are used as anti-epileptic agents, all induce accelerated inactivation of each other and of contraceptive agents. Therefore, oral contraceptives may be rendered ineffective in patients being treated with such transcriptional inducers.
This cartoon is criminally simplified, neglecting a host of other interacting and regulatory proteins, but then this is no class (and I’m no expert) on transcriptional regulation.
| 4.3.1 |
Transcriptional induction of cytochrome P450: Rifampicin bound to the pregnane X receptor |
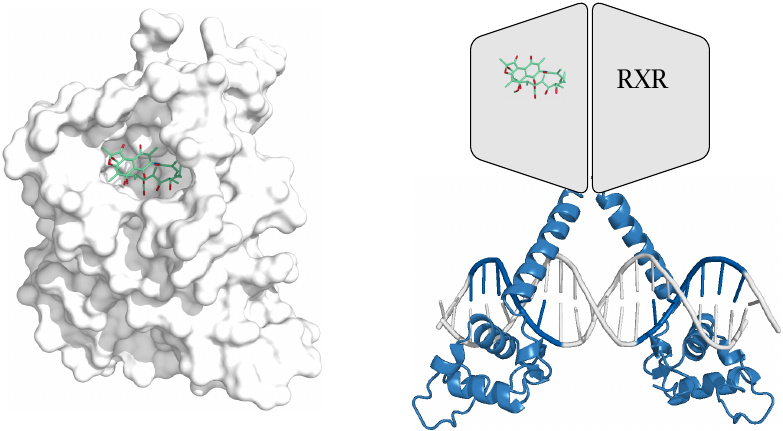
Complete structures of nuclear hormone receptors are apparently not yet available. The structure on the left [22] illustrates rifampicin bound to the drug-binding domain of the pregnane X receptor.26
The ligand-bound PXR forms a heterodimer with another nuclear hormone receptor, the retinoid X receptor (RXR) before binding to the DNA. The partial structure on the right shows the DNA-binding domains of a similar receptor dimer, along with outlines of the two ligand-binding domains. The interaction of a nuclear hormone receptor with DNA is illustrated in slide 7.3.1.
| 4.4 |
Conjugation reactions |
| Functional group | Enzyme | Cosubstrate |
| Glucuronic acid | UDP-glucuronosyltransferases | UDP-glucuronide |
| Sulfate | Sulfotransferases | 3′-Phosphoadenosine-5′- phosphosulfate (PAPS) |
| Glutathione | Glutathione-S-transferases / spontaneous | Free glutathione |
| Acetate | N-acetyltransferases | Acetyl-CoA |
| Methyl | N-, S-, and O-methyl-transferases | S-adenosylmethionine (SAM) |
| Amino acids | Amino acid transferases | Free amino acids/ATP |
Phase II metabolism consists of conjugation reactions. Some of these reactions have already been illustrated; others will be shown below.
| 4.4.1 |
Morphine skips phase I and is conjugated directly |
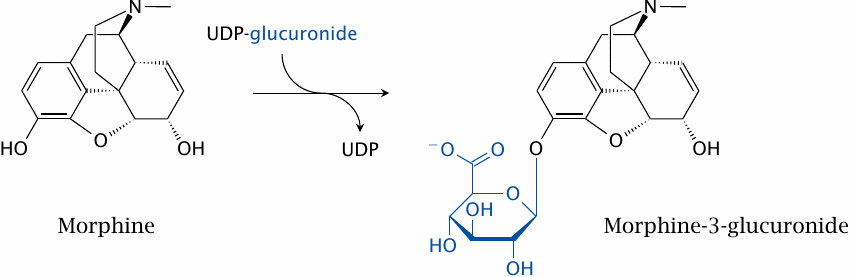
Phase I reactions are not necessary if a drug molecule already contains functional groups suitable for conjugation. An example is morphine, which has two hydroxyl groups. The conjugation of either, or both, with glucuronic acid is sufficient for excretion. The single glucuronide shown in this slide is the major metabolite found in the urine. Interestingly, the other single glucuronide retains pharmacological activity; therefore, even drug conjugates may be active metabolites, although this is less common than with the often more limited modifications introduced in phase I (cf. slide 4.2.2).
In addition to conjugation, morphine may also undergo N-demethylation by cytochrome P450; however, this makes little difference for urinary excretion.
| 4.4.2 |
Epoxides of aromatic hydrocarbons can intercalate and covalently react with DNA |
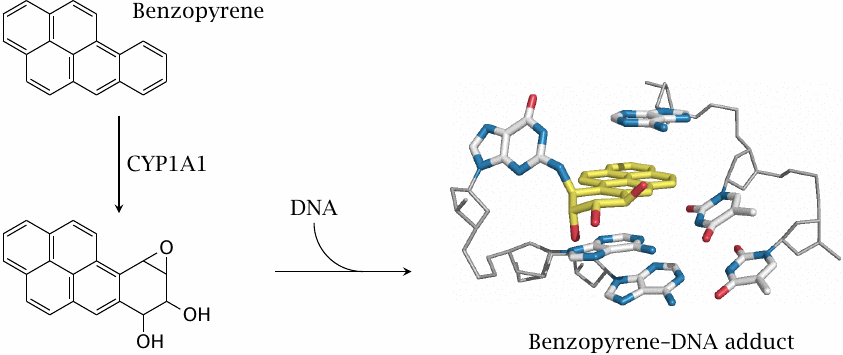
Benzopyrene and related aromatic compounds are contained in tobacco smoke and in industrial emissions. Their activation by cytochrome P450 enzymes, notably subtype 1A1, is an essential step in their carcinogenic action. The study that reported the structure of the DNA adduct shown here, which involves the epoxide on the benzopyrene metabolite and the 2’-amino group of a guanine base, also discusses how this adduct interacts with DNA polymerase to induce mutations [23].
Like CYP3A4, CYP1A1 is also subject to transcriptional induction, but this occurs downstream of a different nuclear receptor (the aromatic hydrocarbon receptor, AHR).
| 4.4.3 |
Enzymatic detoxification of benzopyrene epoxy-derivatives |
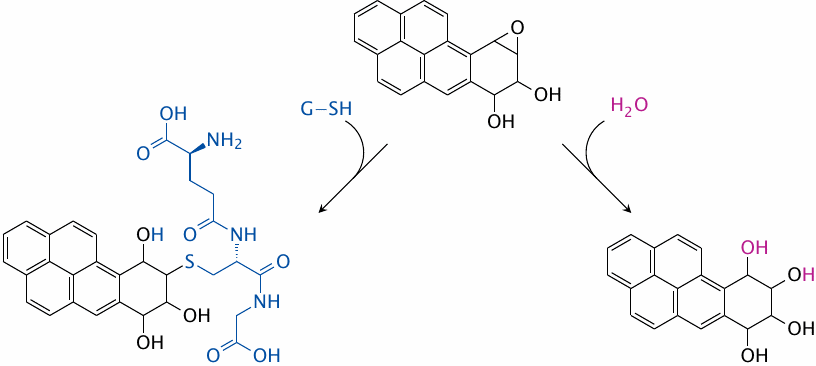
While reactive benzopyrene metabolites are harmful, most such molecules will actually be captured and inactivated before reacting with DNA—otherwise, famous smokers like Winston Churchill and Deng Xiaoping might not have lived long enough to become famous …
The major detoxification pathways are hydrolysis, which is catalyzed by epoxide hydrolase, and glutathione conjugation, which is catalyzed by glutathione-S-transferase (GST).
| 4.4.4 |
Hepatic metabolism of acetaminophen |
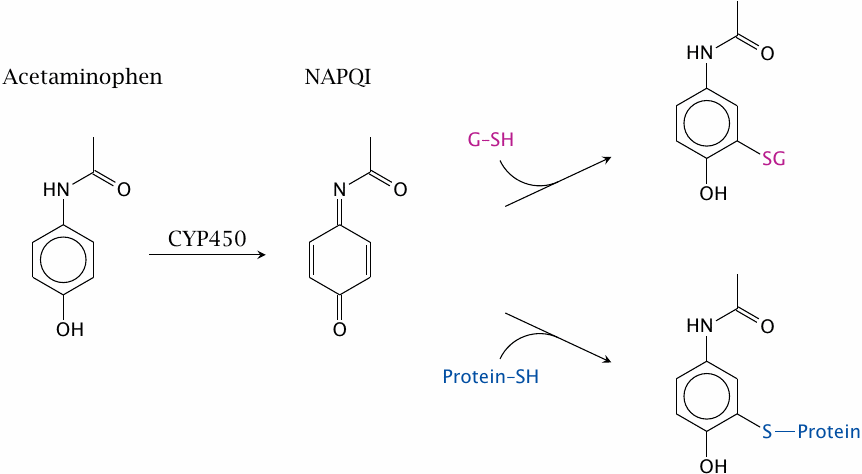
A cytochrome P450-catalyzed oxidative reaction that we haven’t seen so far is the conversion of aromatic rings to quinones. This is exemplified by acetaminophen, which is converted to N-acetyl-p-benzoquinone imine (NAPQI). This metabolite is readily conjugated with glutathione and excreted. If acetaminophen is overdosed, this metabolic pathway can deplete glutathione in the liver cells and thereby cause severe, even fatal, liver toxicity.27
Acetaminophen is used for its inhibitory effect on cyclooxygenase. NAPQI is also formed in the reaction of acetaminophen with cyclooxygenase, and this forms the basis of its inhibitory activity (see slide 9.3.4).
As an alternative to the pathway shown here, acetaminophen can also undergo direct glucuronidation or sulfation at its hydroxyl group [24]. These two competing pathways will mitigate the cytochrome P450-mediated toxicity. The glucuronide is actually the most abundant metabolite found in the urine.
| 4.4.5 |
Activation of canfosfamide by glutathione-S-transferase |
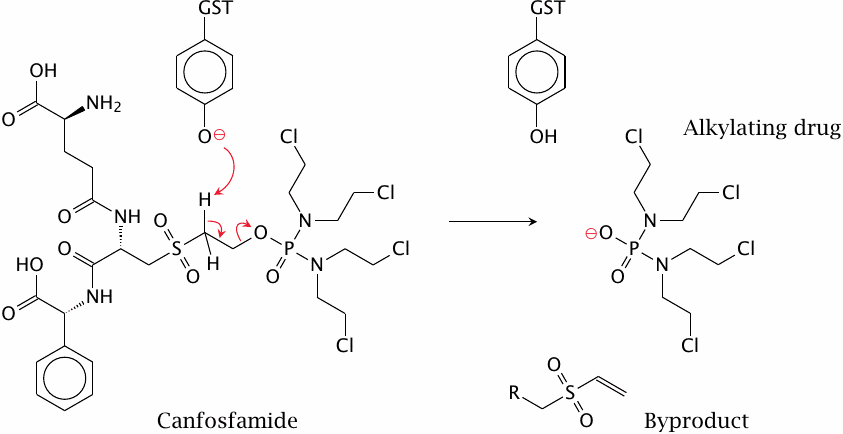
Glutathione reacts readily not only with epoxides but with many other cytotoxic compounds that are reactive toward cellular nucleophiles. Many such compounds are used as anticancer drugs; some examples are shown in slide 12.6.16. During treatment, cancer cells may become resistant to these drugs through mutations that increase the expression of glutathione-S-transferase (GST).
The anticancer drug canfosfamide was designed to turn this resistance mechanism on its head. The molecule, which contains a moiety that resembles glutathione, is cleaved rather than alkylated by GST. It is activated by this cleavage, and it should therefore be more active in tumor cells that overexpress the enzyme.
Canfosfamide is cytotoxic due to its reactive chloroethylamine groups; slide 2.3.5 shows how these groups will react with cellular nucleophiles. These groups are already present in canfosfamide itself, and they are not involved in GST-mediated cleavage; therefore, just how does cleavage by GST increase the drug’s anticancer activity?
While I have seen no experimental evidence for or against it, the following hypothetical explanation seems plausible. The difference in activity between the prodrug and the cleaved product may be due to the extrusion of the prodrug. Glutathione conjugates make good substrates for ABC transporters, which will eject them from the cell. Canfosfamide resembles a glutathione conjugate and thus may also be subject to extrusion. Cleavage by GST releases the reactive moiety of the drug, allowing it to escape extrusion, stay in the cell and exert its cytotoxic effect.
| 4.4.6 |
Acetylation of INH by N-acetyltransferase 2 (NAT 2) |
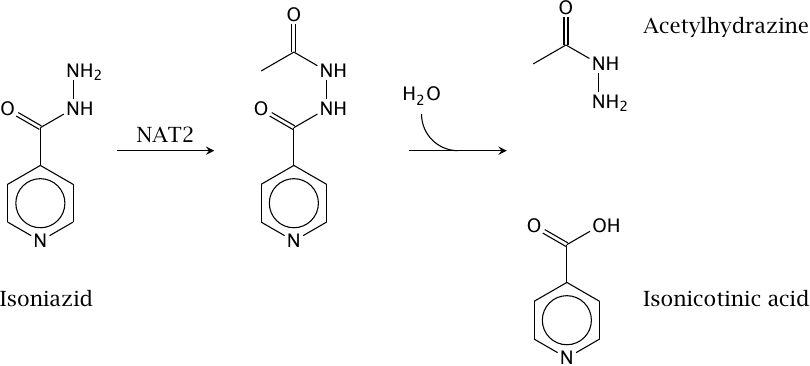
Isoniazid (isonicotinic acid hydrazide, INH) is the classical example of drug metabolism through acetylation. The acetylated product may undergo hydrolysis to release acetylhydrazide, which is reactive and may cause cytotoxicity. Nevertheless, overall, N-acetylation reduces INH toxicity, since metabolites with even greater toxicity would otherwise arise through N-hydroxylation by cytochrome P450 (see below).
In spite of its potential metabolic toxicity, INH is tolerated better than most other tuberculostatic drugs, and it is also the most commonly prescribed one. Its antibacterial mode of action is discussed in slide 11.4.2.
| 4.4.7 |
Bimodal distribution of INH acetylation speed |
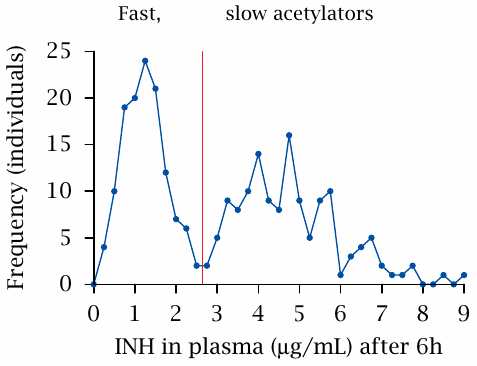
In the depicted experiment, the speed of acetylation was measured in a group of patients. A fixed test dosage (presumably adjusted for body weight) was applied at t=0, and the amount of drug still remaining in the blood plasma after six hours was measured. There clearly are two separate peaks; the two groups represented by these peaks are referred to as fast and slow acetylators, respectively. Figure prepared from original data in [25].
Among Caucasians, about 50% express an inactive NAT2 allele, which causes the slow-acetylator phenotype. The percentage of slow acetylators is lower among Asians (but was higher in a small study done on Kenyans [26]; I don’t know how representative that study is).
Apart from isoniazid, the NAT2 enzyme and its polymorphism also affect the inactivation rates of some other drugs, such as procainamide and hydralazine, which are more likely to cause toxicity in slow acetylators. NAT2 has also been implicated in the susceptibility to bladder cancer caused by aromatic amines, which in Europeans was found to correlate with slow acetylator status. Surprisingly, the same correlation was not observed in Chinese [27]. I have not seen an explanation for this discrepancy.
| 4.4.8 |
Metabolic activation of arylamine carcinogens |
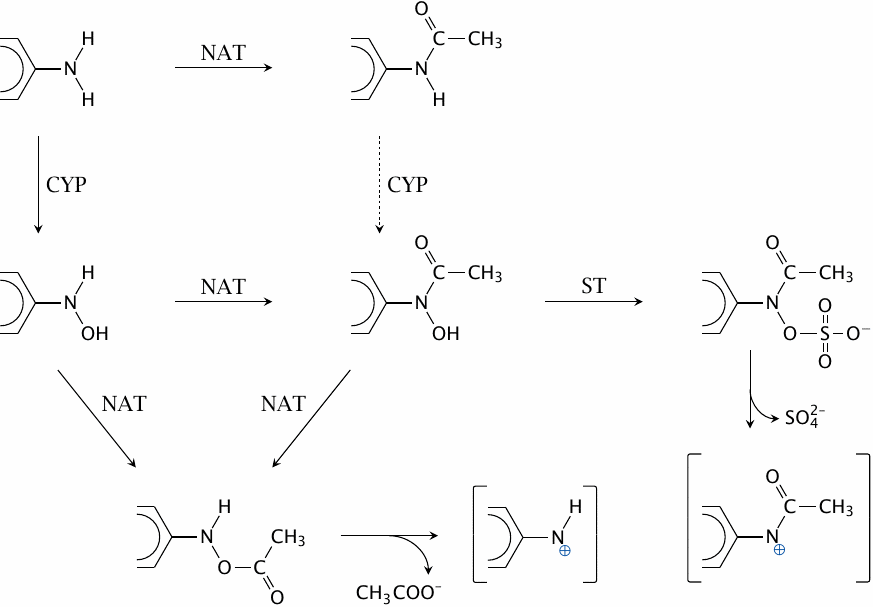
N-Acetyltransferases (NAT), cytochrome P450 (CYP) and sulfotransferases (ST) cooperate in the metabolic activation of arylamine carcinogens such as benzidine or 2-naphthylamine. The acetoxy and sulfohydroxamate products decay spontaneously to reactive electrophiles, which can then react covalently with cellular macromolecules, including DNA.
The activation is most efficient if CYP acts before NAT; acetylation of arylamines therefore affords partial protection from carcinogenic activation. This likely accounts for the lower susceptibility of fast acetylators to arylamine-induced tumors. Figure drawn after a scheme shown in [28].
| 4.4.9 |
Glutamine conjugation of phenylacetate |
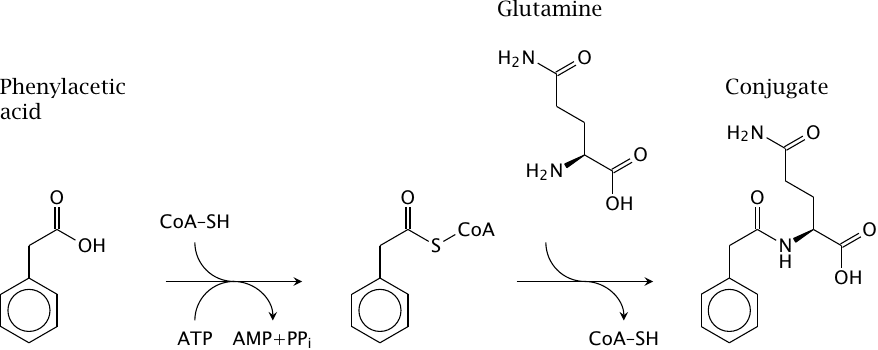
Glutamine, glycine and taurine can be conjugated to various xenobiotics that are organic acids; the figure shows glutamine conjugation of phenylacetic acid as an example.
Glycine and glutamine conjugation are exploited in an unusually ingenious manner for alternate pathway therapy in enzyme defects of the urea cycle (see slide 10.2.6).
| 4.5 |
Reductive drug metabolism |
- important functional groups in substrates: nitro, azo, sulfoxide, quinones
- diverse enzymology
- “incidental”—most enzymes that cause reductive drug metabolism primarily serve other roles in metabolism
- some reductive reactions can occur without enzyme catalysis
Reductive drug metabolism belongs to phase I. It is less regular than the oxidative and conjugative pathways covered so far, and also somewhat less common than the most prominent oxidative and conjugation pathways.
Among the various enzymes involved in reductive metabolism, there are a few surprises, such as cytochrome P450 enzymes and xanthine oxidase, which on occasion may transfer electrons to acceptors other than oxygen. We will look at some examples but make no attempt at exhaustive coverage.
| 4.5.1 |
Reductive activation of sulindac by thioredoxin |
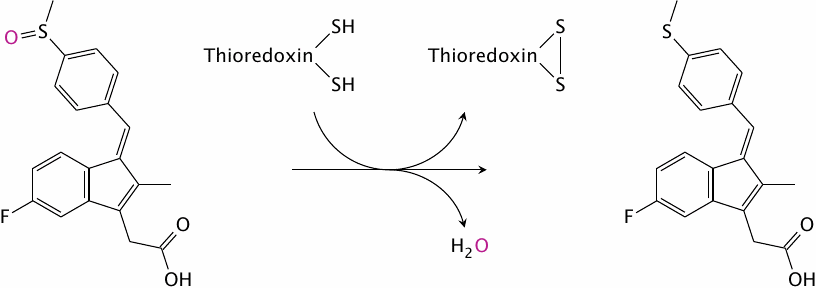
Sulindac is a prodrug that is activated by reductive metabolism, which is carried out by the reducing enzyme thioredoxin; the resulting thioether is a cyclooxygenase inhibitor. After reduction, the drug is excreted into the bile and then reabsorbed from the small intestine. This entero-hepatic cycling gives the drug a fairly long half life (~16 hours).
| 4.5.2 |
Redox-active ingredients of Vicia faba |
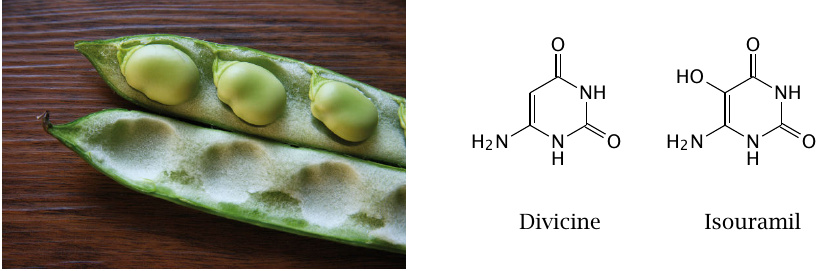
The broad bean (Vicia faba) contains isouramil and several other compounds isouramil and divicine that undergo repeated oxidation and reduction in the body, and in the process burn up reduction equivalents.
| 4.5.3 |
Redox cycling of isouramil |
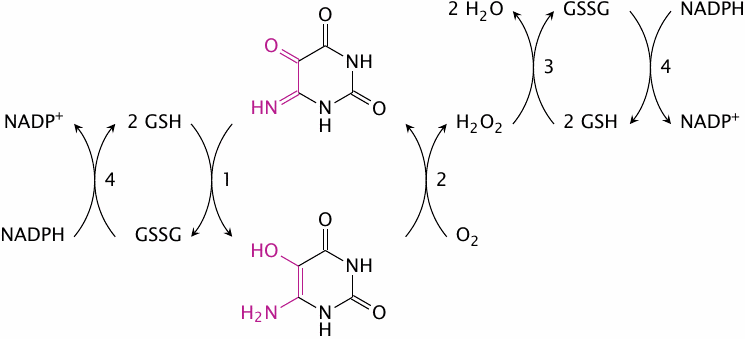
This scheme outlines the redox cycle that is induced by isouramil. Reactions 1 and 2 occur spontaneously, without the requirement for any enzymatic catalysis. The H2O2 formed in reaction 2 is then reduced by glutathione peroxidase. The glutathione disulfide formed in reactions 1 and 3 is reduced at the expense of NADPH in reaction 4 by glutathione reductase. The NADPH, in turn, must be regenerated by other pathways.
| 4.5.4 |
Glucose-6-phosphate dehydrogenase deficiency leads to favism |
- Most patients are healthy most of the time—hemolytic crises triggered by drugs or food ingredients that cause redox cycling
- Manifest in red blood cells because these cells lack protein synthesis—no replacement of deficient enzyme molecules during the lifetime of the cell
- Affords partial protection against malaria—similar to sickle cell anemia and other hemoglobinopathias
In red blood cells, the only pathway that provides NADPH is the hexose monophosphate shunt. The first enzyme in this pathway is glucose-6-phosphate dehydrogenase. Mutations that lead to a reduced activity of this enzyme limit the supply of NADPH and, therefore, the capacity of the cell to cope with redox cycles such as the one created by isouramil.
The toxicity of reactive oxygen species is mediated by the peroxidation of membrane lipids, which destroys the cell membrane—the red blood cells disintegrate. The occurrence of such episodes of hemolytic anemia upon ingestion of broad beans is called favism.
In contrast to red blood cells, nucleated cells can replace inactive enzyme molecules. Furthermore, they also contain mitochondria and can replenish cytosolic NADPH using mitochondrial NADH. Therefore, the enzyme defect is not manifest in nucleated cells.
| 4.5.5 |
Redox cycling of 5-hydroxyprimaquine |
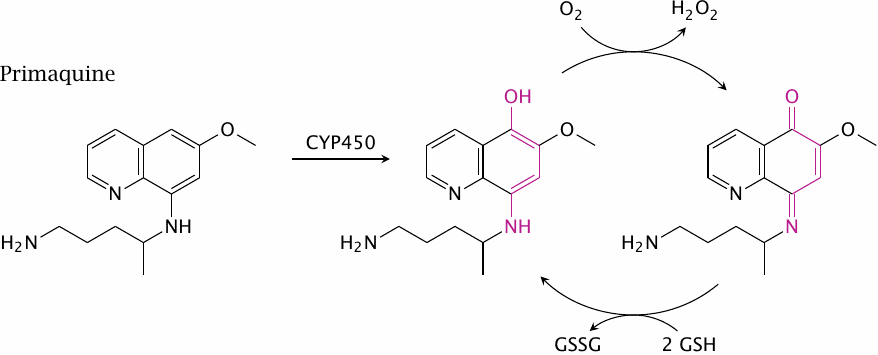
Like sickle-cell anemia and several other hemoglobinopathias, glucose-6-phosphate dehydrogenase deficiency is found more frequently among people indigenous to countries in which malaria is common. This is something to keep in mind when treating malaria patients with primaquine, which after hydroxylation to 5-hydroxyprimaquine by cytochrome P450 sets up a redox cycle analogous to the one induced isouramil (only shown in part in this slide). Primaquine therefore may induce hemolytic anemia in these patients. Several other drugs can do the same.
| 4.5.6 |
The anticancer prodrug CB1954 bound to quinone reductase 2 |
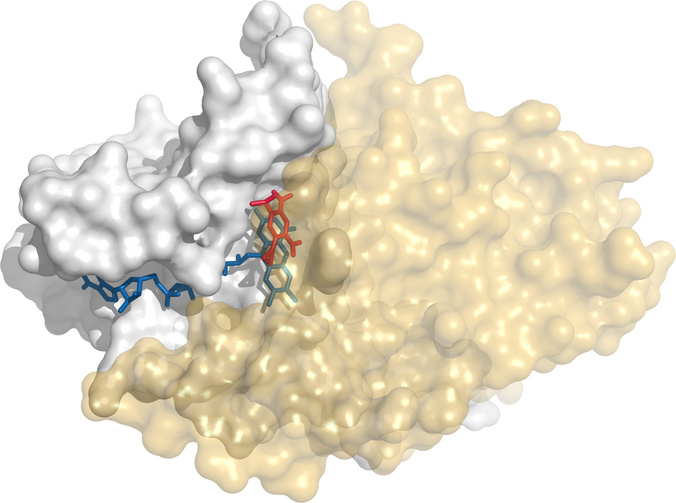
CB1954 is an experimental anticancer prodrug that is activated by reductive metabolism. In this structure [29], CB1954 (red) is shown bound to diaphorase, or quinone reductase 2. The cosubstrate NAD and the coenzyme FAD are shown in red. One of the enzyme’s subunits is rendered transparent to make the substrate visible.
| 4.5.7 |
Two-step activation of the anticancer prodrug CB1954 |
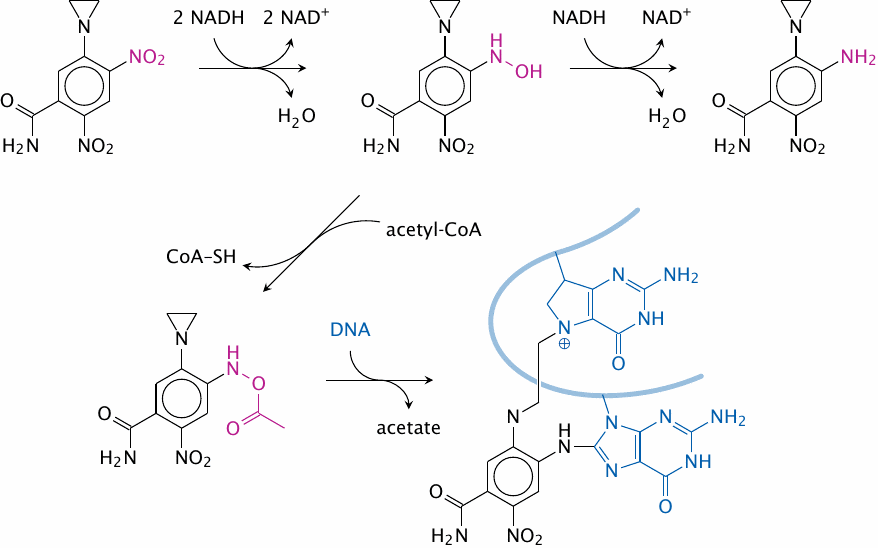
CB1954 is not a quinone, from which we can infer that the name ‘quinone reductase’ for the enzyme in question is too narrow. Indeed, the same enzyme also reduces methemoglobin and cytochrome b5; considering such a broad range of endogenous substrates, it is not surprising that it accepts drugs in a fairly non-specific manner also.
The compound undergoes nitro reduction, followed by acetylation. The resulting acetoxy group is reactive toward DNA, as is the aziridine group already contained in the parent molecule. Such bivalent reagents—or multivalent ones such as canfosfamide, see above—can introduce crosslinks into DNA molecules, which are difficult for the cell to repair and therefore inflict much more effective cytotoxic damage than univalent reagents. Many anticancer drugs are, or give rise to, multivalent reagents (see section 12.6).
For reasons that are not well understood, the cytoplasm of tumor cells tends to be more reducing than that of healthy cells; so, drug activation by reduction is a way to preferentially target tumor cells. Note though that reductive activation also applies to some established anticancer drugs, such as mitomycin C or bleomycin. These two drugs are nevertheless quite toxic, so we should probably not expect particularly low toxicity from reductively activated synthetic drugs such as CB1954.