| 15 |
Drug discovery |
| 15.1 |
Stages of drug discovery |
-
Target molecule
- selection
- validation
-
Candidate compounds
- acquisition
- screening
Biochemical and physiological discoveries suggest macromolecules that might make worthwhile targets for treating diseases. This is an important step toward new drugs, but only the first one. The subsequent steps that must be taken are the subject of this chapter.
Drug development is labor-intensive and costly, so it is important to experimentally ascertain whether a drug with the intended activity on our chosen target would indeed produce the expected physiological outcome. This stage of development is referred to as target validation.
The choice of the experimental model for target validation depends on the intended physiological effect. Some effects, for example cytotoxicity, can readily be observed in cell cultures; on the other hand, for physiological functions such as pain or blood pressure, animal experiments will be required. If the drug under consideration is supposed to be an inhibitor, its effect can often be modeled by genetic knockout of the target or by RNA interference (see slide 13.3.6). On the other hand, if the intention is to activate the target, it should in principle often be possible to model this by overexpression (cf. slide 2.6.1); however, this approach does not seem to be commonly used in practice.
If the target has been experimentally validated, candidate compounds must be obtained and screened. Major pharmaceutical companies have large libraries (collections) of compounds that may be screened over and over against novel, unrelated targets. Alternatively, custom libraries may be synthesized and can be structurally focused around some existing agonist or antagonist of the target molecule.
The screening of large numbers of compounds—a recent study on a specific type of voltage-gated K+ channels tested as many as 650,000 compounds [137]— requires simple and robust high-throughput assays. If detailed structural information on the macromolecular target is available, it may be preferable to perform the initial screening in silico, that is, to use molecular docking software to examine the binding of real or virtual compounds to the target.
Once lead compounds have been identified that act on the target in the intended manner, they must also be tested with other macromolecules that are related to the target; the entirety of such data for a given compound is its receptor profile. Lead compounds must also be tested against antitargets, that is, macromolecules that are frequently involved in drug toxicity. One important antitarget are cardiac hERG potassium channels, whose inhibition by drugs gives rise to cardiac arrhythmias (see section 6.6).
| 15.1.1 |
Chemical structures of subtype-selective glutamate receptor ligands |
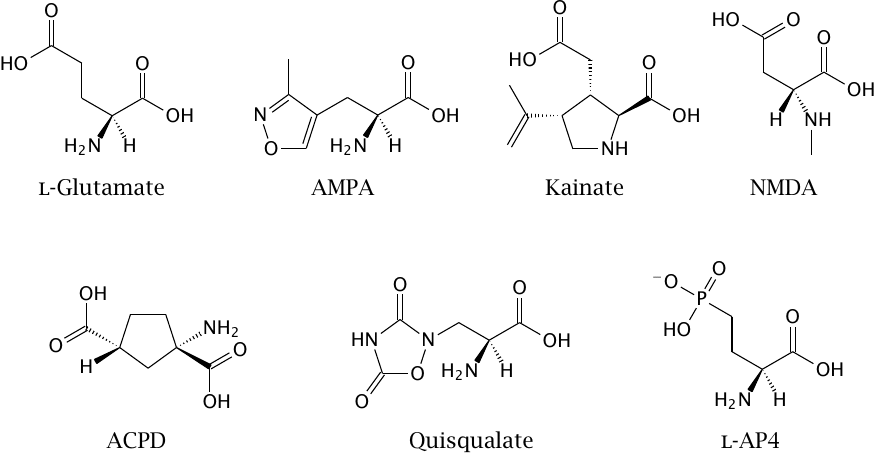
While a drug development project will usually start with a hypothesis that is based on the state of the art in biochemistry and physiology, one must nevertheless be ready to reevaluate this hypothesis as further experimental information accrues along the way. A common case in point is the discovery of pharmacologically distinct subtypes of receptors that at the outset were assumed to be homogeneous.
The molecules shown in this slide were prepared as candidate agonists for glutamate receptors in the central nervous system. As it turned out, these synthetic ligands did not all act the same way, but instead activated different subsets of glutamate receptors, the existence of some of which was only thereby discovered.
AMPA and quisqualate activate the same subtype of ionotropic glutamate receptors. Quisqualate also activates group I metabotropic glutamate receptors, which belong to the GPCR type. NMDA and kainate, respectively, activate two other ionotropic receptor subtypes. ACPD is an agonist of group I and II metabotropic glutamate receptors. l-AP4 is an agonist of group III metabotropic receptors (see section 6.11).
| 15.2 |
Sources of candidate compounds |
- Synthetic libraries
- Natural compounds
- Semisynthesis
- Gene technology
Candidate compounds can be obtained from various sources. Natural compounds or synthetic strategies can be used alone or in combination; the latter approach is termed semisynthesis. Gene technology is a promising development but not yet a major source for compounds in practice.
| 15.2.1 |
Combinatorial synthesis: the Ugi reaction |
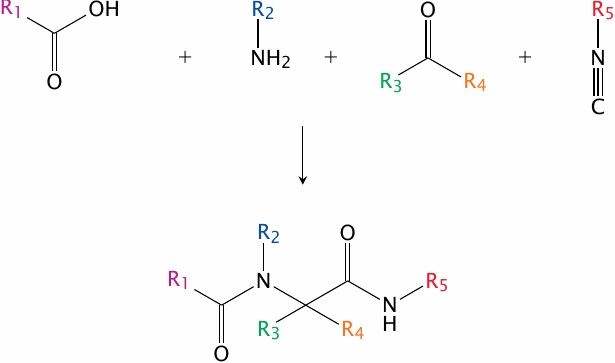
One important strategy for developing large compound libraries is referred to as combinatorial synthesis. In this approach, modular reactants that share similar reactive groups but differ in their side chains are combined in many permutations, all of which are processed in parallel, often with the help of robotic systems.
An example of a combinatorial synthetic strategy is the Ugi reaction. Here, a carboxylic acid, an amine, a ketone or aldehyde and an isonitrile condense to form a bis-amide. Through combinatorial variation of the functional groups R1–R5, many different compounds can be prepared in parallel. An application of the Ugi reaction for the combinatorial synthesis of analogues of the antibiotic muraymycin is described in [138].
| 15.2.2 |
Semisynthesis of penicillins |
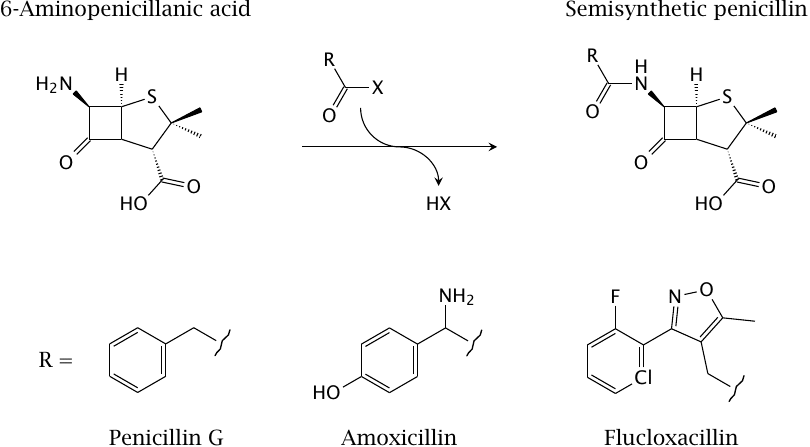
The penicillins are good examples for the semisynthetic variation of natural compounds. The starting compound, 6-aminopenicillanic acid, is obtained from fermenter cultures of the fungus Penicillium notatum, and is chemically acylated at its unique amino group.
| 15.2.3 |
Semisynthesis of cephalosporins |
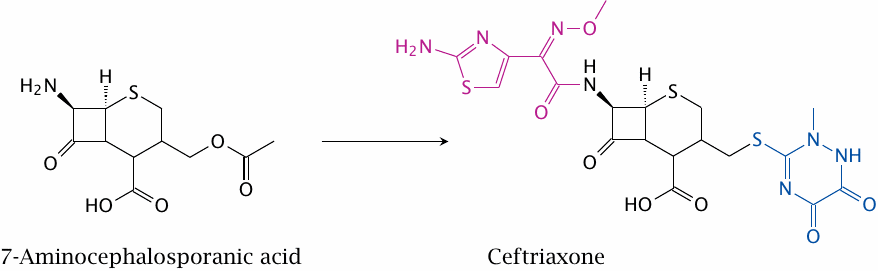
Ceftriaxone (shown here) and other cephalosporins (cf. slide 11.4.10) are derived through semisynthesis from 7-aminocephalosporanic acid. Here, two variable substituents are attached to the natural compound nucleus. More complex, multi-step chemistries are regularly utilized on a wide variety of natural products.
| 15.2.4 |
Biosynthesis of polyketides |
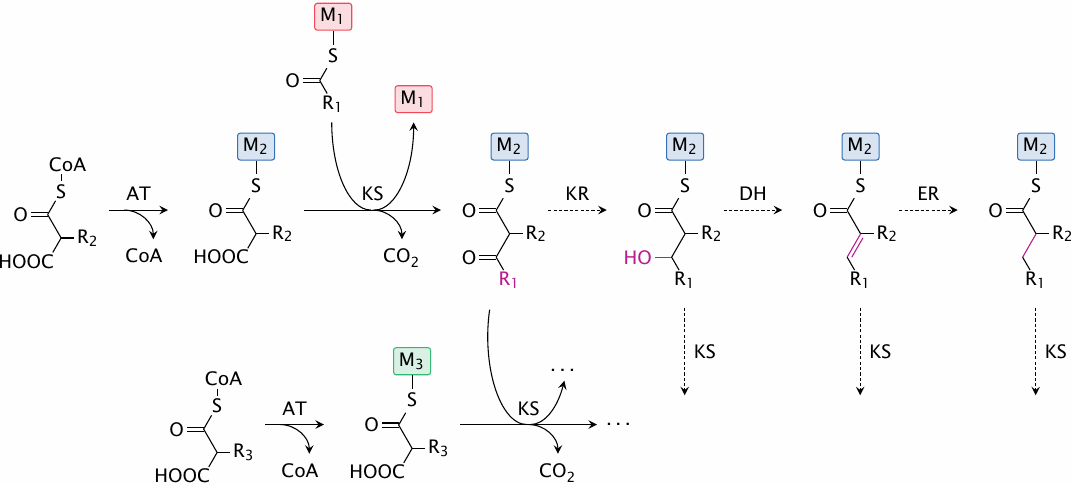
Polyketides are natural compounds that are produced, in considerable variety, by Streptomyces species and other microorganisms. The biosynthesis of polyketides is chemically similar to that of fatty acids, with the following differences:
- In contrast to fatty acids, which are assembled through stepwise attachment of a single precursor (malonyl-CoA), polyketides are contain various organic acids with different side chains. Each polyketide has its specific composition and sequence of side chains.
- While the β-carbon of each successively added C2 subunit is always completely reduced in fatty acid synthesis, in polyketides the reduction of β-carbons may be complete or partial.
Polyketide synthases are large molecules, structured like assembly lines, in which a given nascent polyketide molecule travels from one module to the next. Each module selects and attaches a specific substrate acid, and then optionally reduces it; the level of reduction is determined by the presence or absence of ketoreductase, dehydratase, and enoylreductase activities in the module in question.
| 15.2.5 |
Structure of native 6-deoxyerythronolide B synthase |
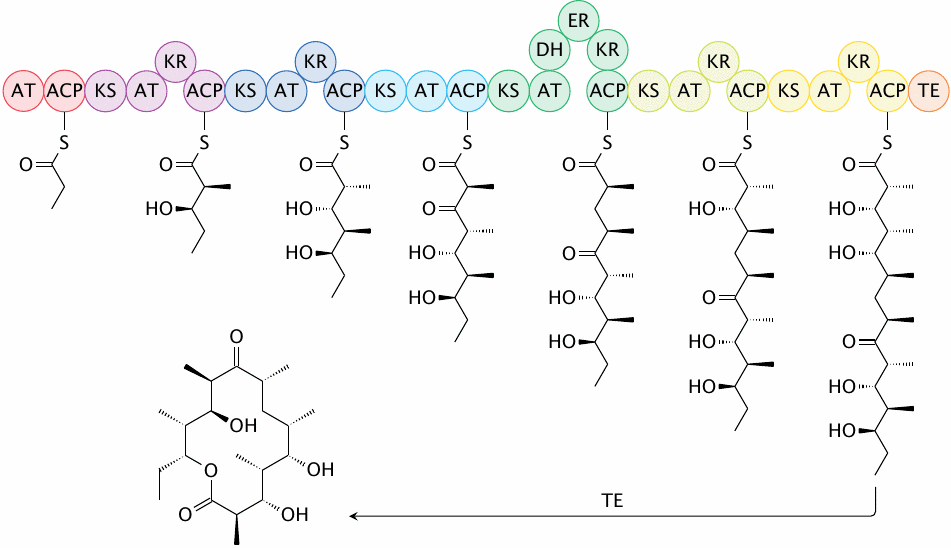
An example polyketide is 6-deoxyerythronolide B, which is a precursor of the antibiotic erythromycin. The synthase that makes this compound has six separate modules, five of which contain one or more reducing enzyme activities, which will cause partial or complete reduction of the corresponding β-carbon atom. The thioesterase (TE) that follows the last module cyclizes and releases the product.
The sequence of the various modules and the presence of reductase domains within these modules completely determine the structure of the product. Recombinant DNA technology can be used to replace individual domains and thereby create novel polyketide molecules.
| 15.2.6 |
Two compounds produced by engineered variants of 6-deoxyerythronolide B synthase |
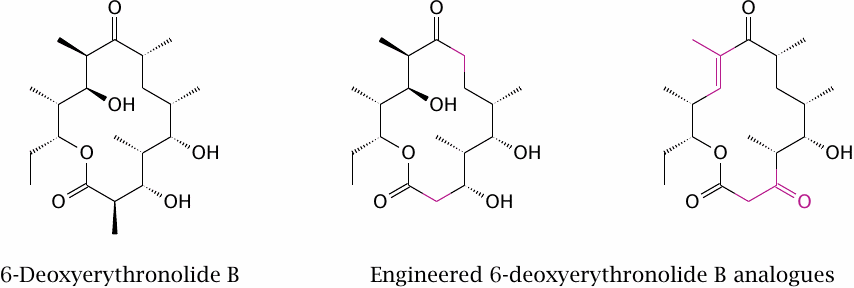
The two engineered variants of 6-deoxyerythronolide B were obtained by replacing several modules of the cognate synthase with corresponding ones from the rapamycin polyketide synthase. Data from [139].
While this approach is intriguing and has the potential to provide novel compounds with intricate structures in a very scalable manner, the scope of structural variation that can be realized in this way will often be more limited than with organic synthesis or semisynthesis.
| 15.3 |
Compound screening |
| Experimental approach | Typical applications |
| Activity assays on purified target proteins | Enzymes |
| Cell-based activity assays | GPCRs, ion channels |
| Computational screening | Targets with available 3D structure |
| Phenotypic screening | Cytotoxic activity |
The best approach for testing the effect of candidate compounds varies with the nature of the target molecule. Enzymes are typically active in solution and can therefore be tested as purified molecules, which minimizes interference by other macromolecules. In contrast, the functions of GPCRs and ion channels can only be observed in cells or model membranes, and cell-based assays are most commonly used. With both in vitro and cell-based assays, absorbance- or fluorescence-based readouts are preferable, since they lend themselves to automation and high throughputs.
Computational screening is best used with target molecules for which detailed structural information is available. While it sometimes is involved and time-consuming, one great advantage of this approach is that the candidate compounds don’t have to physically exist before the screen; actual synthetic effort can be focused on those candidates that score well in silico.
Phenotypic screening is different from the other entries here, since it is performed without any specific target molecule; instead, the desired physiological outcome, such as the killing of bacterial or cancer cells, is measured directly. Many anticancer and antimicrobial drugs have emerged from phenotypic screens.
| 15.3.1 |
Peptide deformylase and Met aminopeptidase |
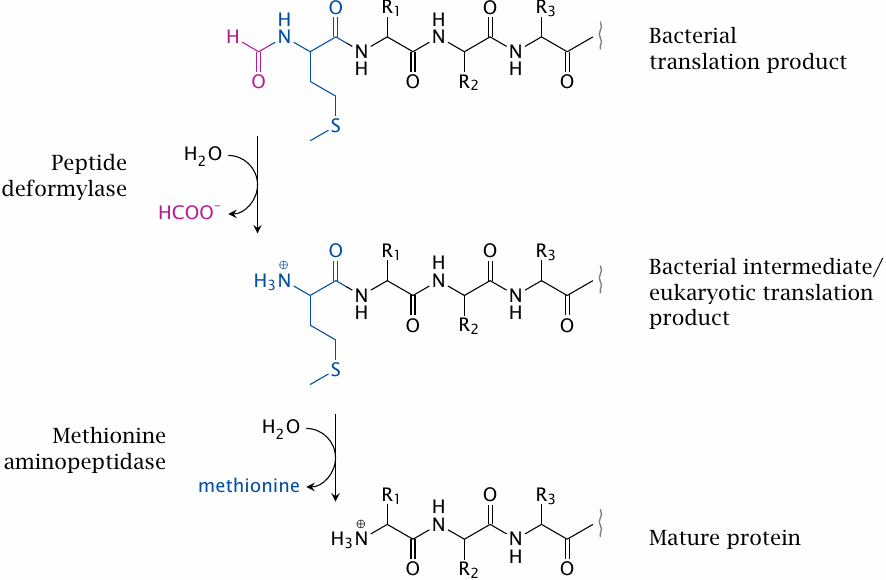
Peptide deformylase and methionine aminopeptidase can serve as examples as drug targets amenable to in vitro screening. Both enzymes function in the posttranslational processing of protein N-termini. In bacterial proteins, but not in eukaryotic cells, the N-terminal methionine residue is N-formylated. The formyl group is removed by peptide deformylase, which accordingly is a candidate target for antibacterial drugs.
In both bacterial and eukaryotic cells, the N-terminal methionine residue is cleaved by methionine aminopeptidase; many proteins will not become functional when this methionine residue is not removed. Accordingly, inhibitors of the enzyme may be of use in both antibacterial and anticancer chemotherapy. The search for antibacterial inhibitors of methionine aminopeptidase brought some surprises that illustrate potential pitfalls of the seemingly straightforward approach of screening with purified enzymes in vitro.
| 15.3.2 |
In vitro screening of Met aminopeptidase inhibitors |
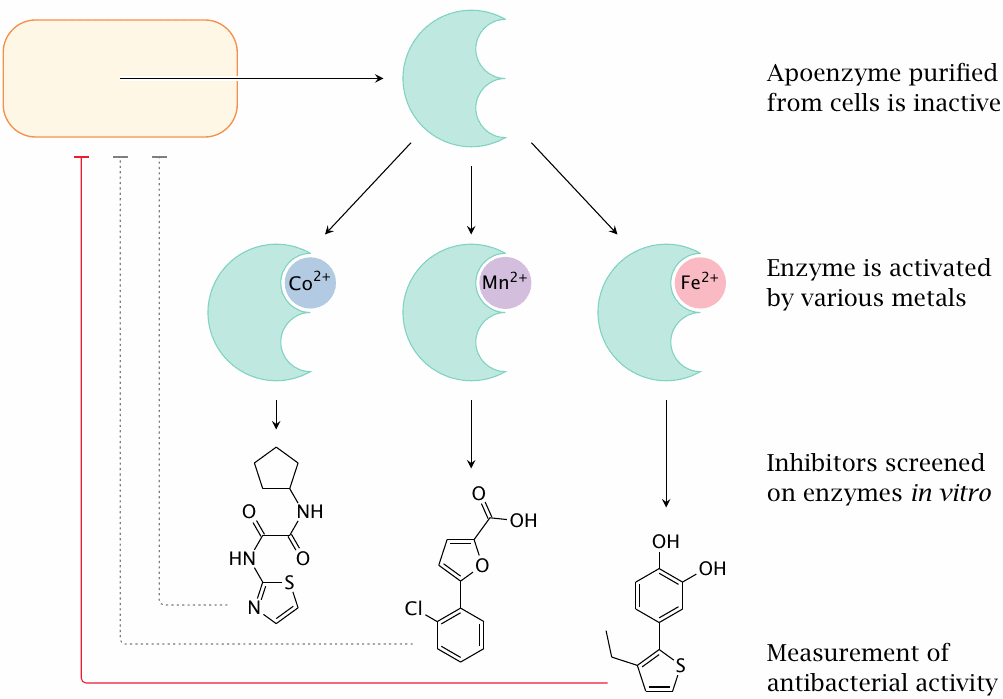
Purification of the protein from E. coli yields the apoenzyme, which is inactive. It can be activated by saturating it with several different metal ions, which suggested the exact nature of the metal to not be critical. Accordingly, several differently metallated preparations of the enzyme were used, somewhat arbitrarily, to screen enzyme inhibitors.
As it turned out, however, the different screens produced different inhibitors, and only the ones screened with Fe++-saturated enzyme turned out to have antibacterial activity. This finding suggests that only this form of the enzyme is relevant in vivo[140]. This example shows the importance of fully characterizing the target’s behavior in vivo.
| 15.3.3 |
A non-covalent yet irreversible enzyme inhibitor |
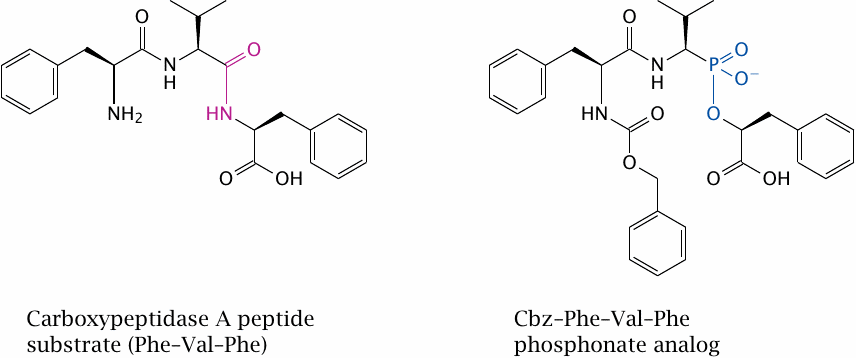
When screening drugs that bind to a specific target, how high should one aim in terms of affinity? The higher the affinity, the lower the required dosage of a drug; practical considerations indicate that a nanomolar affinity is desirable. While this may seem ambitious, even higher affinity can be achieved on occasion, as is the case with the inhibitor shown in this slide.
The target molecule in question is carboxypeptidase A. The structure on the left shows a tripeptide substrate of this enzyme; the one on the right is that of the inhibitor Cbz-Phe-Val-Phe phosphonate. The time constant for dissociation of this femtomolar inhibitor exceeds the lifetime of the enzyme, which means that inhibition is practically irreversible [141].
| 15.3.4 |
A generic assay for GPCR activation |
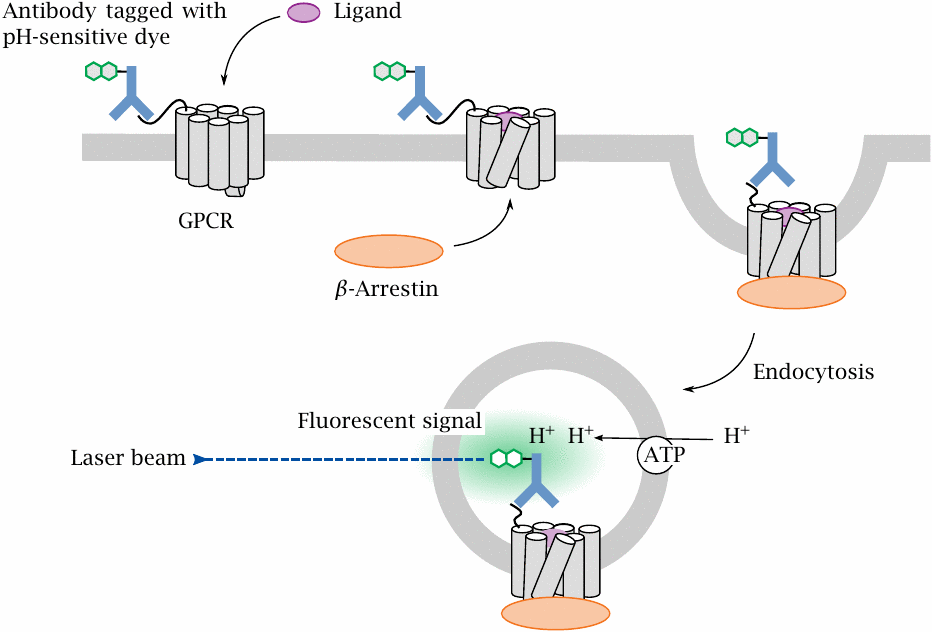
GPCR function depends on several other cellular components, and therefore can only be screened with cell-based assays. Many screens rely on some specific signal that is triggered downstream of the activation of the specfic GPCR in question, or at least of a specific G protein. In contrast, the CypHer 5 assay illustrated here responds to endocytosis of the receptor itself, which in turn is triggered by receptor activation. The assay is therefore very general.
In preparation for the assay, the GPCR of interest is overexpressed in cells, with its N-terminus fused to an antigenic peptide that is then tagged with a cognate antibody carrying a pH-sensitive fluorescent dye. When a ligand activates the receptor, the receptor is phosphorylated and binds β-arrestin, which in turn triggers receptor endocytosis. When the endosome containing the labeled receptor is acidified by cellular proton pumps, the fluorescent dye is protonated, making it brightly fluorescent.
| 15.3.5 |
A cell-based fluorescence assay of membrane depolarization |
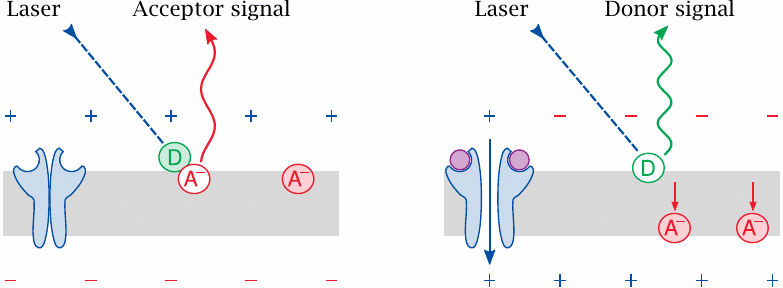
A technically challenging kind of drug target to screen in a high-throughput format are ion channels. The cell-based assay shown here uses fluorescence to detect membrane depolarization downstream of channel activation.
In preparation for the assay, the cell membranes are loaded with two lipophilic fluorescent dyes, which are spectrally matched for fluorescence resonance energy transfer (FRET) but differ in net charge and in transverse mobility.
At the resting membrane potential, the two dyes stay close to one another, and excitation of the donor dye causes FRET and long-wave emission from the acceptor dye. If, however, a ligand activates the channel and reverses the membrane potential, the dyes become separated; the donor will then emit directly within its own, characteristic wavelength range.
| 15.3.6 |
A fluorescence assay of Ca++ influx |
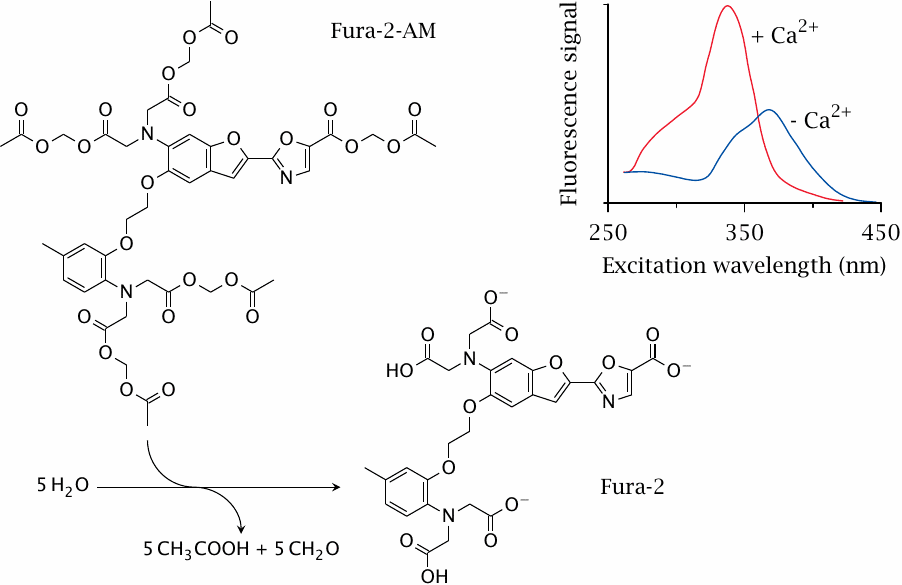
The cytosolic Ca++ level is normally low, but it can rise due to influx of extracellular Ca++ through CaV channels or to efflux from the ER downstream of GPCRs that couple to phospholipase C (see slide 5.3.2). To measure cytosolic calcium signals downstream of a drug target, cells can be loaded with a resorption ester of the fluorescent dye Fura 2. Cleavage by cellular esterases exposes the dye’s two Ca++-chelating functional groups.
When intracellular calcium increases, it binds to the dye, which leads to a strongly increased fluorescence signal upon excitation at 340 nm.
| 15.3.7 |
An in silico docking experiment |
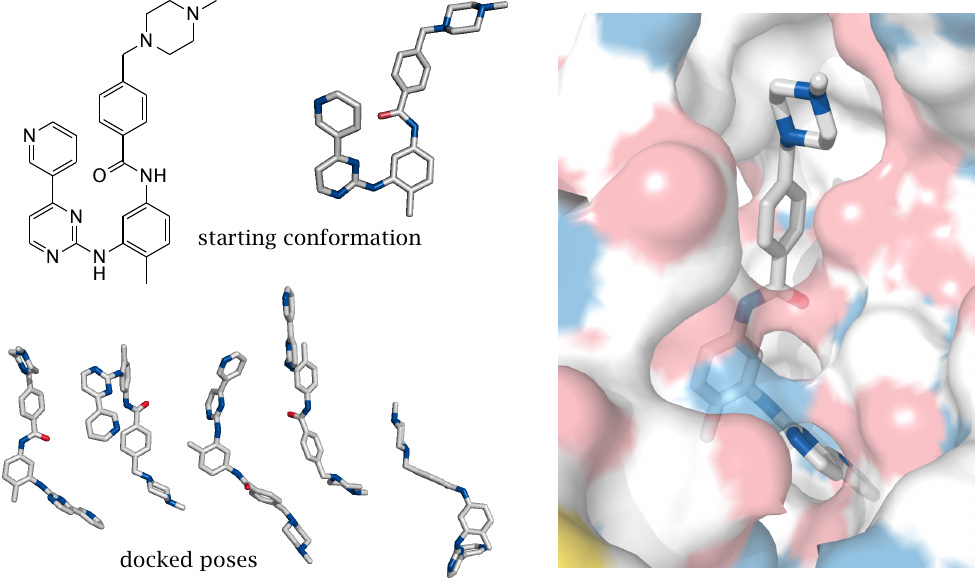
The model experiment illustrated here was carried out using the freely available program AutoDock Vina [142] and followed a detailed tutorial that can be found on that program’s website.
The protein tyrosine kinase inhibitor imatinib was docked into the active site of the abl oncoprotein tyrosine kinase (cf. slide 12.5.4). The starting conformation of imatinib was given to the docking program, which was allowed to rotate the single bonds of the drug molecule in order to optimize its fit to the enzyme (whose structure was treated as invariant).
Below the starting conformation, several docked poses, that is, energetically favorable ligand orientations, are shown. The most energetically favorable pose is almost indistinguishable from the one obtained by crystallography.
| 15.3.8 |
Electrostatic potential mapped onto the electronic density for acetaminophen |
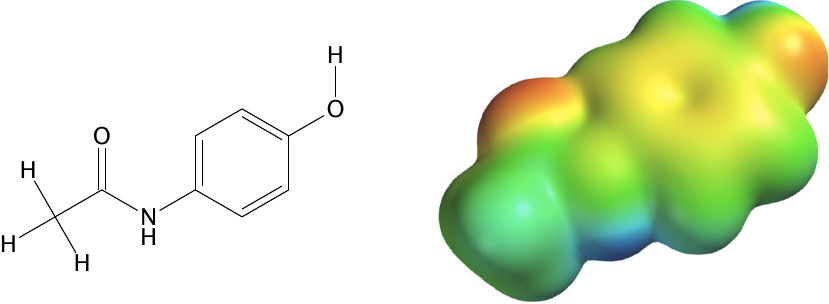
Docking programs such as AutoDock Vina that are designed for initial screens of large numbers of compounds treat molecules in a substantially simplified manner; among other things, all molecular charges are assigned to discrete point coordinates. For more accurate prediction of binding parameters, it is necessary to consider the electronic structure at higher resolution.
This slide illustrates the distribution of electrostatic charges across the van der Waals contour of the drug acetaminophen. The calculation was performed using the commercial software Spartan 08 (Wavefunction Inc, USA). The calculation was performed at the RHF/6-31G⋆ level of theory.
| 15.3.9 |
Hypothetical pharmacophore for inhibitors of ATP:L-Methionine S-Adenosyltransferase |
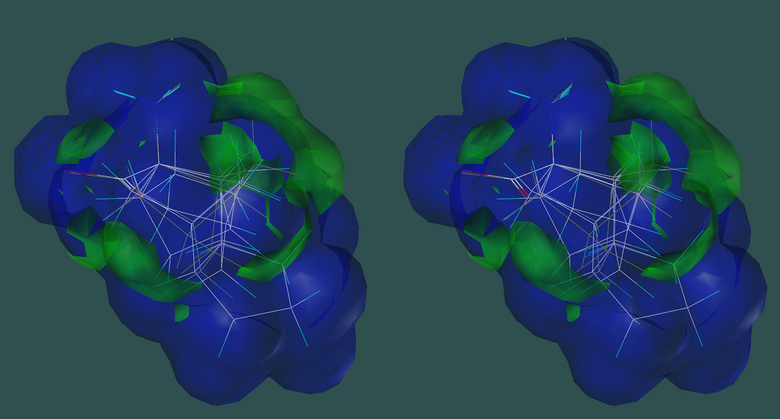
While actual crystal structures of drug targets are highly desirable—and nowadays often available—for computational screening, another viable approach is based on the pharmacophore concept. In this approach, molecular features of the ligand that enhance or detract from the desirable effect on the target are determined by experimentally characterizing and comparing a set of reference compounds. The structures of the compounds are aligned in space, and each molecular feature that enhances or detracts from the desired effect is expressed by its magnitude, direction, and nature of binding force. The pharmacophore is the consensus set of favorable features. Once the pharmacophore has been established, further compounds can be compared to it by computational analysis.
The slide illustrates a pharmacophore for the enzyme ATP:L-Methionine S-adenosyltransferase, which regenerates the methyl group donor S-adenosylmethionine. In the construction of this pharmacophore, six different derivatives of 1-aminocyclopentane-1-carboxylic acid were tested for their ability to displace methionine from the enzyme. White lines indicate the bonds of the aligned compounds. The volume that envelopes all active compounds is outlined in blue; the volumes of substituents that prevent binding extend out and are indicated in green. Based on data in [143].
Sybyl8.1 (Tripos, Inc, USA) software was used to produce this cross-eyed stereo figure.