| 1 |
Introduction |
| 1.1 |
What is biochemical pharmacology? |
What is it?
- pharmacology, but with a focus on how drugs work, not on whether we should take them before or after dinner
- fascinating—you will love it, or double your money back
What is it not?
- just molecular pharmacology—physiological context is important, too
- a claim that we completely understand the biochemical action modes of all practically useful drugs—we don’t
Let me repeat the last two points—the action modes of many drugs are incompletely understood, and physiology is important. I will try to cover the key aspects of physiology as far as they are needed to understand the course material, but you may find it useful to consult an actual physiology text on the side from time to time.
| 1.1.1 |
On drugs and poisons: Paracelsus’ maxim |

We will now consider the question: “What are drugs?” Our first witness will be the gentleman shown in this slide. In his day—the 16th century—Paracelsus acquired a reputation not just as a physician, but also for his fondness of a good drink; rumor has it that this latter inclination was useful to him in the discovery of his famous maxim.
As you know, many drugs turn poisonous if overdosed, and many poisonous natural compounds may become useful as drugs when properly diluted. That holds for both legal and illegal drugs; we will consider examples of both classes in this course.
| 1.1.2 |
A very small drug particle and a very large one |
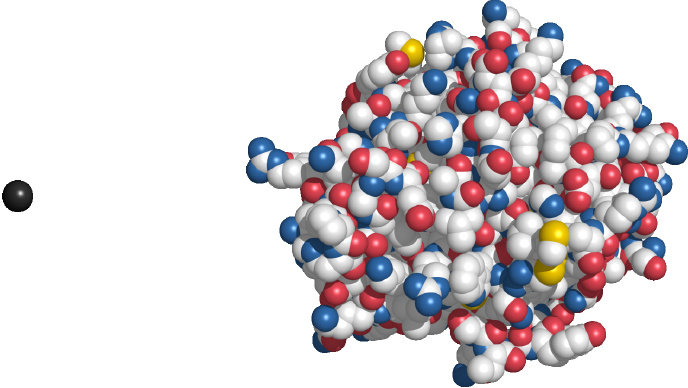
Lithium is the smallest drug particle—it is not even a molecule but just a single atom. It is also an example of a drug whose mode of action is still unknown, despite many decades of research; various biochemical effects have been described, such as the inhibition of certain protein kinases, but no single effect has been conclusively linked to its antidepressant activity in humans.1
Urokinase is a protein and as such is much larger than most other drug molecules. It is used to dissolve intravascular blood clots in order to restore perfusion in acute myocardial infarction and stroke.
Note that neither lithium nor urokinase is a very typical drug; some more typical ones are shown in the next slide.
| 1.1.3 |
Some drug molecules of more typical size |
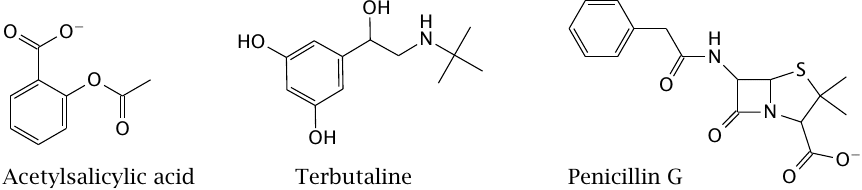
Most drugs are organic molecules with molecular weights of no more than 500 Da, such as the examples shown here. This rule of thumb holds for drugs of very different functional classes.2 As you may know, acetylsalicylic acid inhibits cyclooxygenase, and penicillin inhibits bacterial cell wall synthesis; terbutaline activates β2-adrenergic receptors. We will discuss all these drug actions in more detail in later chapters.
Organic drug molecules may be natural compounds or obtained through organic synthesis. Some drugs are obtained semisynthetically, that is, through the synthetic modification of natural compounds; for example, penicillin G and other penicillin derivatives are obtained through semisynthesis.
| 1.2 |
Drugs and drug targets |
Practically all drugs—with the exception of those that act osmotically; see below—must bind to target molecules in order to exercise their effects. Most drug targets are proteins.
| 1.2.1 |
Functional classes of protein drug targets |
- Enzymes
- Hormone and neurotransmitter receptors
- Ion channels
- Membrane transporters
- Cytoskeletal proteins
As we work through the example drugs in this course, you will notice that most of the drug targets fall into one of the above categories.
| 1.2.2 |
Non-protein drug targets |
- DNA: alkylating anti-tumor drugs
- RNA: anti-ribosomal antibiotics, antisense oligonucleotides
- Lipid membranes: antibiotics (amphotericin B, polymyxin); gaseous narcotics, alcohol?
- Free space, or rather no target at all: osmolytes
Drug targets other than proteins are less common. DNA is important with anticancer drugs. The RNA of bacterial ribosomes is targeted by various classes of antibiotics. It is noteworthy, however, that many regulatory effects of RNA have only recently been discovered and appreciated. In experimental research, it has become quite common to inhibit the expression of specific genes at the level of mRNA, and in a few cases this strategy has already found clinical application. This is discussed in more detail in chapter 13.
Drugs that target lipid membranes are again most clearly exemplified by some antibiotics, which recognize specific lipids that occur in microbial cell membranes but not mammalian ones. The accumulation of alcohol and of gaseous narcotics in the cell membranes of neurons may contribute to the effects of these compounds; the long-standing question as to what extent these effects arise from direct interaction with membrane proteins as opposed to membrane lipids has not yet been fully resolved.
Osmotically active drugs are used in order to influence the distribution of fluids between compartments. An important application is the use of dextran or hydroxyethyl-starch as blood plasma expanders, that is, to replace lost blood volume. The osmotic activity of such macromolecules counteracts the hydrostatic pressure difference that exists across the capillary walls of the circulation. If lost blood were replaced with physiological saline solution instead, this fluid would rapidly leak out into the tissues due to the intracapillary pressure.3
While plasma expanders are clinically important, they are not very interesting from a biochemical point of view, and therefore we will not consider them any further.
| 1.2.3 |
Histamine receptor antagonists |
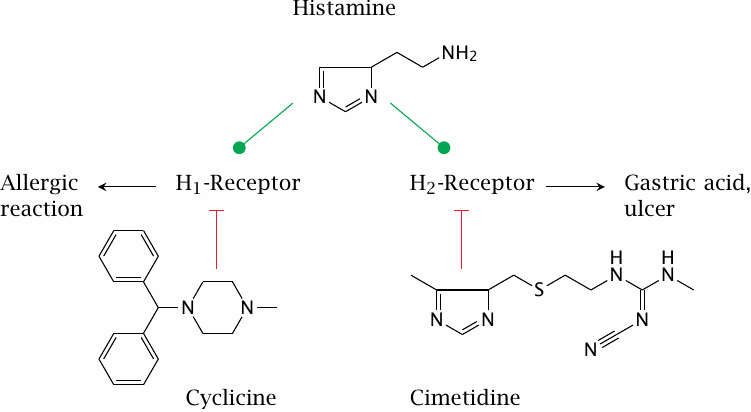
Among the hormone and neurotransmitter receptors, a particularly prominent class are the G protein-coupled receptors (GPCRs). These receptors are found in the cytoplasmic membranes of all kinds of cells and respond to a large number of different ligands. It is often said that approximately 50 % of all clinically used drugs target G protein-coupled receptors. We will devote all of Chapter 5 to this important receptor class; here, we will briefly look at histamine receptors and angiotensin receptors as examples.
Histamine receptors occur in several subtypes. While all subtypes are activated by the same physiological ligand—that is, histamine—they may differ in their susceptibility to inhibition by receptor antagonists. For example, cyclizine inhibits H1-receptors, whereas cimetidine selectively blocks H2-receptors. Subtype-selective drugs are often useful in pharmacotherapy; for example, H1-selective inhibitors can be used to treat allergies without interfering with acid secretion, and H2-selective drugs can be used to treat gastric hyperacidity without causing drowsiness, which is a common side effect of H1-receptor blockade.
While cyclizine has only remote structural similarity to histamine, the resemblance is more obvious with cimetidine. This similarity is not coincidental.
| 1.2.4 |
The development of H2-receptor blockers |
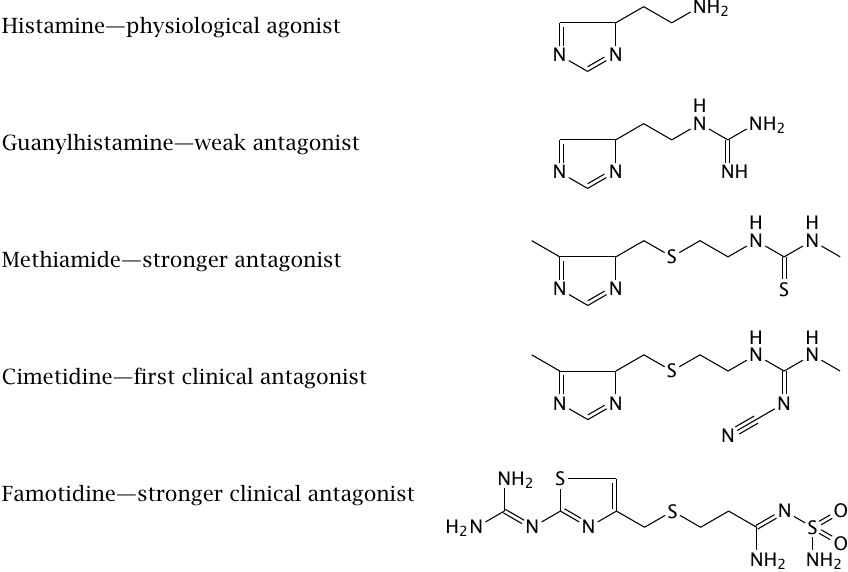
The development of cimetidine began in the 1960s. The histamine receptors had not been molecularly characterized at the time; all that was known about them was that they existed and were activated by histamine, and that some of them were involved in the secretion of gastric acid. Gastric acid was known to be a factor in the causation of gastric ulcers.4 The guiding idea was to modify the structure of histamine so as to turn it from an activator into an inhibitor, which could then be used to treat the ulcers.
This plan proved remarkably successful. The slide illustrates intermediate stages of the development process. Guanylhistamine was the first derivative that retained affinity for the receptor but inhibited it. Further modification yielded cimetidine, which improved upon the receptor affinity of guanylhistamine and became the first clinically used H2-receptor antagonist, serving as the mainstay of ulcer therapy for more than a decade. Even more avidly binding antagonists, such as famotidine, can be administered at dosages of just a few milligrams per day, rather than the several hundreds of milligrams that were required with cimetidine.
| 1.2.5 |
Angiotensin: Proteolytic release from angiotensinogen, and mode of action |
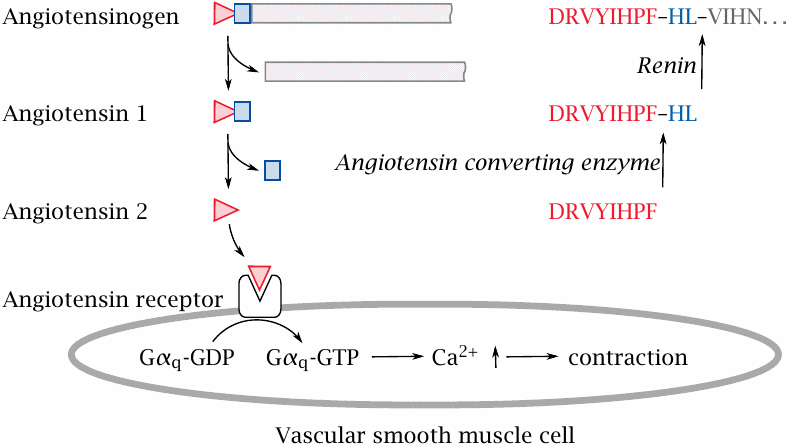
Angiotensin 2 is a peptide mediator that plays an important role in the control of blood pressure. It binds to a GPCR at the surface of vascular smooth muscle cells. An intracellular signaling cascade then induces contraction of the cells, which causes constriction of the blood vessels and increased blood pressure. Inhibition of angiotensin 2 action is an important principle in the treatment of hypertension.
Angiotensin is released in two successive proteolytic steps from angiotensinogen, a plasma protein. Accordingly, angiotensin action can be inhibited by preventing either its proteolytic release or its binding to the receptor.
| 1.2.6 |
Two inhibitors of proteolytic angiotensin release |
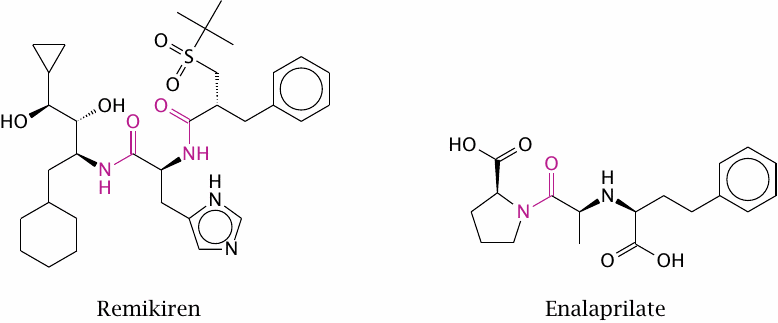
Remikiren inhibits the protease renin, which catalyzes the first proteolytic cleavage of angiotensin, whereas enalaprilate inhibits angiotensin-converting enzyme, which performs the second cleavage step. Both drug molecules contain peptide bonds, which are highlighted. Peptide bonds are subject to cleavage by proteases and peptidases in the digestive tract. In the case of remikiren, this prevents oral drug application, since the drug is destroyed by these enzymes before it can be taken up. In contrast, the single peptide bond in enalaprilate has a greater degree of steric protection, and enalaprilate can therefore be used orally.
It is interesting to compare the structure of these protease inhibitors to the peptide sequences of the angiotensinogen substrate near the respective cleavage sites (see preceding slide).
| 1.2.7 |
Sequence of saralasin, a peptide inhibitor of the angiotensin 2 receptor |
| Peptide | Sequence |
| Angiotensin | Asp-Arg-Val-Tyr-Ile-His-Pro-Phe |
| Saralasin | Sar-Arg-Val-Tyr-Val-His-Pro-Ala |
Saralasin is a synthetic peptide whose sequence is a variation of that of angiotensin. Sarcosine (Sar) is N-methyl-glycine.
Saralasin cannot be applied orally, because as a peptide it gets degraded by proteases and peptidases in the intestine. Even when applied intravenously, saralasin is fairly rapidly degraded by peptidases in the blood plasma, much like angiotensin itself. While drugs with such short lifetimes are impractical for routine application, they can occasionally be useful. For example, a hypertensive crisis is a dangerous condition that is characterized by spontaneous spikes in blood pressure that are very high yet of short duration. In this situation, drugs with short lifetimes make it possible to rapidly increase and decrease the antihypertensive effect so as to match these spontaneous changes in blood pressure. A longer-acting drug might overshoot when the blood pressure spike subsides spontaneously and drive the blood pressure below a safe target value.
| 1.2.8 |
Non-peptide ligands of peptide receptors |
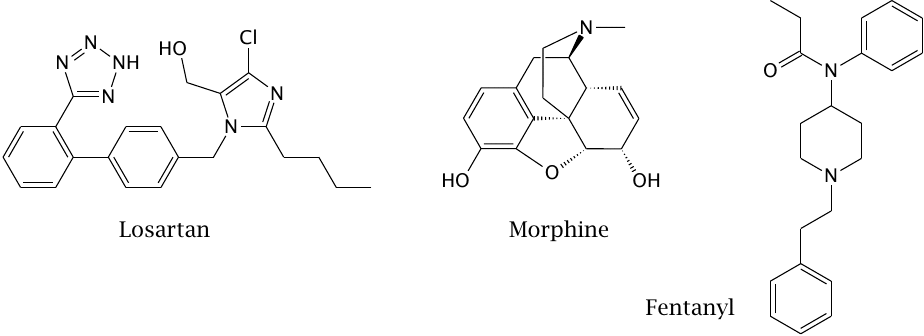
Like cimetidine, saralasin was developed through the stepwise modification of a physiological receptor agonist (that is, an activating ligand) so as to turn it into an antagonist. However, as we have seen, peptide molecules are inherently unstable in vivo. The question therefore arises whether peptide receptors can be controlled with more stable non-peptide ligands. The drugs in this slide illustrate that this is indeed possible.
Morphine is a natural compound that binds and activates opioid receptors, a class of GPCRs activated by endogenous peptide agonists called endorphins. Morphine was the first non-peptide ligand of a peptide receptor to be identified. Fentanyl is a synthetic opioid receptor agonist that has a considerably simpler structure.
Losartan is a synthetic inhibitor of the angiotensin 2 receptor. In contrast to saralasin, it is not susceptible to degradation in the intestinal tract, and it therefore can be orally applied. Losartan itself and several similar drugs are widely used in the treatment of hypertension.5
| 1.3 |
Drug discovery and development |
In the development of cimetidine and of saralasin, the physiological receptor agonists were used as lead compounds. Starting with a lead compound is an important approach to drug development, but not the only one. Some drugs have been found through phenotypic screening, without any previous knowledge or assumptions about suitable molecular targets. This approach is alive and well, although nowadays it is often possible to start with a known target molecule, and often even with the chosen target’s complete molecular structure.
| 1.3.1 |
Arsphenamine, the first modern antibacterial drug |
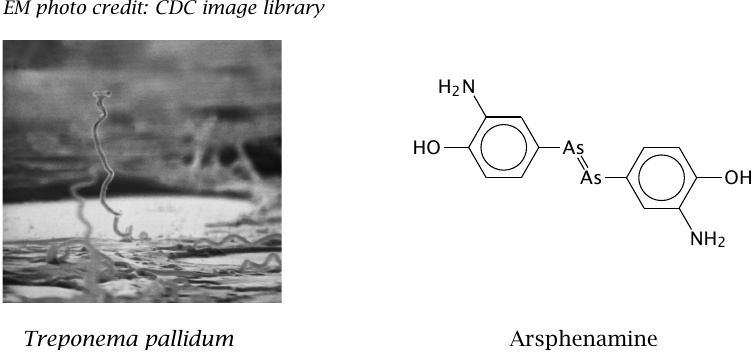
The antibacterial activity of arsphenamine was discovered in the early 20th century by Paul Ehrlich and Sachahiro Hata, who screened more than 600 compounds for activity against syphilis. Since Treponema pallidum, the bacterium that causes syphilis, cannot be grown in vitro, the screening had to be done in animal experiments.
Arsphenamine can be considered an early example of drug development by brute-force phenotypic screening. It was used for several decades in the treatment of syphilis, until penicillin became available.
| 1.3.2 |
Paul Ehrlich, the discoverer of arsphenamine and originator of the receptor concept |

A 200 Deutschmark bill (no longer valid; image from wikimedia) with Paul Ehrlich’s portrait and the structure of arsphenamine. Ehrlich’s research institute was located in the city of Frankfurt; the background of the image shows historical buildings from the city center.6
| 1.3.3 |
Drug discovery by brute force: sulfamidochrysoidine |
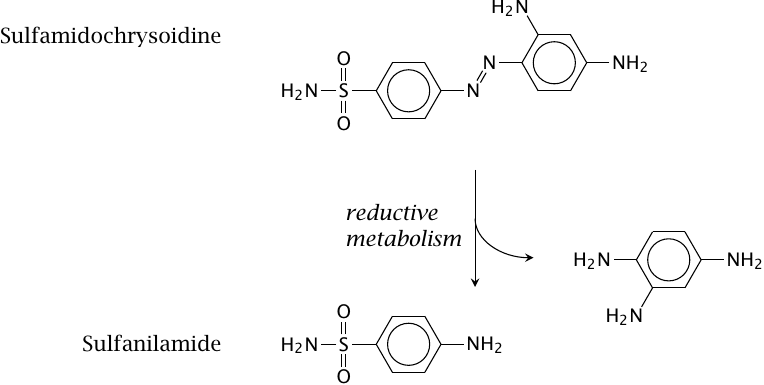
Like arsphenamine, sulfamidochrysoidine emerged from an early large-scale drug screening program. While arsphenamine was active against syphilis only, sulfamidochrysoidine was the first drug with activity against a broad spectrum of bacterial pathogens.
Sulfamidochrysoidine itself is inactive in vitro; if you add it to a bacterial culture, nothing happens. The active component, sulfanilamide, is released from sulfamidochrysoidine through reductive drug metabolism in vivo.7 Therefore, as with arsphenamine, the antibacterial effect could only be observed in animal experiments.
Sulfanilamide and other sulfonamides are antimetabolites of folic acid synthesis. Most bacteria synthesize folate themselves and are unable to take it up from the environment. In contrast, folic acid is a vitamin in humans, which means that it is acquired from the diet, and therefore inhibition of folic acid synthesis doesn’t affect us. Therefore, sulfonamides can be used to treat infections in humans.
| 1.3.4 |
Natural compounds and semisynthetic derivatives |
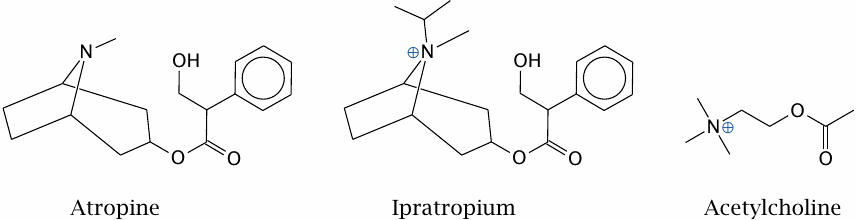
Atropine is an alkaloid found in the plant Atropa belladonna (deadly nightshade). It acts as an antagonist of certain types of acetylcholine receptors. This has a range of effects on physiological regulation, such as acceleration of heartbeat and relaxation of bronchi and of pupils. At higher dosages, atropine also interferes with brain function. Atropine is a very good example of the drug–poison duality observed by Paracelsus.
Ipratropium is a semisynthetic derivative of atropine. Attachment of an isopropyl group to the nitrogen turns the latter into a quaternary amine. This gives the molecule a permanent charge, which prevents it from penetrating the brain as easily as atropine does, and therefore reduces side effects. We will consider the basis of this effect in Chapter 3.
| 1.3.5 |
Protein structure-based drug discovery: HIV protease bound to its inhibitor saquinavir |
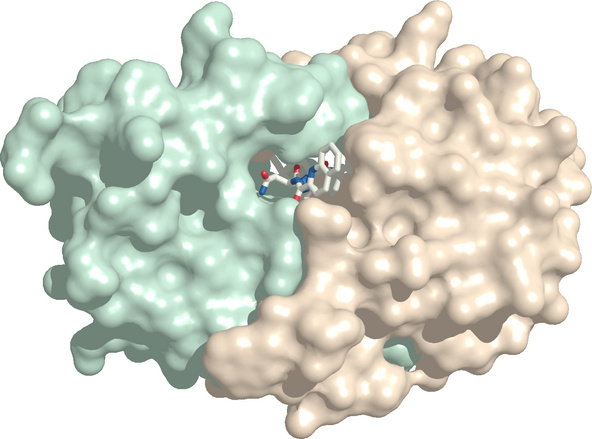
The HIV protease inhibitor saquinavir illustrates the modern paradigm of protein structure-based drug development.
Like several other viruses, HIV expresses its proteins in the form of a single polyprotein precursor, which is then proteolytically cleaved into the individual proteins. This cleavage is performed by the HIV protease, a viral protease which releases first itself and subsequently the other domains (see slide 11.10.7). If this cleavage is prevented, the proteins will remain trapped and not become functional; HIV protease therefore is essential for the maturation of virus particles. Accordingly, inhibition of the protease is an effective strategy for treating HIV infection.
The protease consists of two subunits that enclose the active site. Once the structure of the protease had been determined, inhibitors such as saquinavir could be designed that fill and block the active site. Structure rendered from 1fb7.pdb.
HIV infections always must be treated with a combination of different drugs in order to prevent the emergence of resistance. Protease inhibitors are a crucial part of such drug combinations, and their availability has greatly extended the life expectancy of HIV patients.
| 1.3.6 |
Structure of saquinavir, and its conformation in the active site of HIV protease |
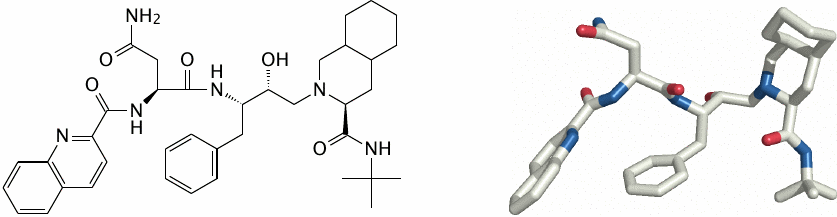
This slide shows the conformation of saquinavir when bound to HIV protease. It is taken from the same structure file as the slide above.
| 1.3.7 |
Drug discovery by accident (1): From a letter by Reverend Edmund Stone to the Royal Society, 1763 |
Among the many useful discoveries, which this age hath made, there are very few which, better deserve the attention of the public than what I am going to lay before your Lordship.
There is a bark of an English tree, which I have found by experience to be a powerful adstringent, and very efficacious in curing anguish and intermitting disorders.
About six years ago, I accidentally tasted it, and was surprised at its extraordinary bitterness … As this tree delights in a moist or wet soil, where agues chiefly abound, the general maxim, that many natural maladies carry their cures along with them, or that their remedies lie not far from their causes, was so apposite to this particular case, that I could not help applying it; and that this might be the intention of Providence here, I must own had some little weight with me …[2]
I continue to be puzzled by the kind of “accident” that might have caused the Reverend to involuntarily taste a piece of willow bark. After all, bicycles or roller blades were not in common use yet, so exactly how did he manage to hurl himself teeth-first into a willow tree? Nevertheless, for the rest of us, the Reverend’s accident certainly proved most fortunate.
| 1.3.8 |
The active ingredient of willow bark, and its more widely known derivative |
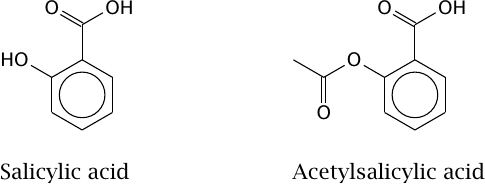
Both salicylic acid and acetylsalicylic acid inhibit cyclooxygenase, a key enzyme in the synthesis of pro-inflammatory prostaglandins and related mediators (see Chapter 9). While salicylic acid is a noncovalent inhibitor, acetylsalicylic acid inhibits the enzyme covalently, and thus irreversibly; this explains its longer lasting action.
The discovery of acetylsalicylic acid, too, was due to luck more than design. Felix Hoffmann, a chemist with Bayer AG, started playing with the structure of salicylic acid, trying to spare his father, who had been prescribed the drug, the intensely bitter taste that had already been noted by Reverend Stone. One of the modifications he tried was acetylation. The resulting drug turned out to have a significantly stronger and longer lasting effect than salicylic acid itself. We will examine the reason for this improved activity in Section 9.5.
| 1.3.9 |
Drug discovery by accident (2): The discovery of penicillin |
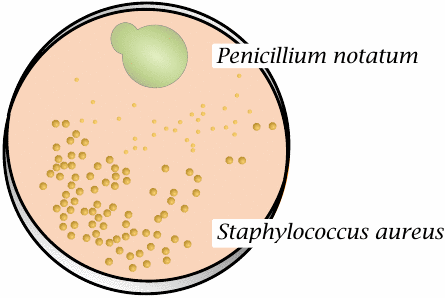
This is a sketch of the original petri dish on which Alexander Fleming first observed the production of penicillin by the mold Penicillium notatum; it is drawn after a photograph in Fleming’s paper [3]. The antibacterial effect of penicillin can be observed by the lysis of Staphylococcus aureus colonies near the mold colony.
The accident that gave rise to this discovery was the mold contamination—normally, microbiologists try to protect their cultures from such contamination. It is to Fleming’s credit that he immediately recognized the importance of this observation. However, his report was not the first of its kind; several others preceded it, and even the term “antibiosis” predates Fleming’s discovery [4].8
| 1.3.10 |
Not all bacteria are susceptible to penicillin |
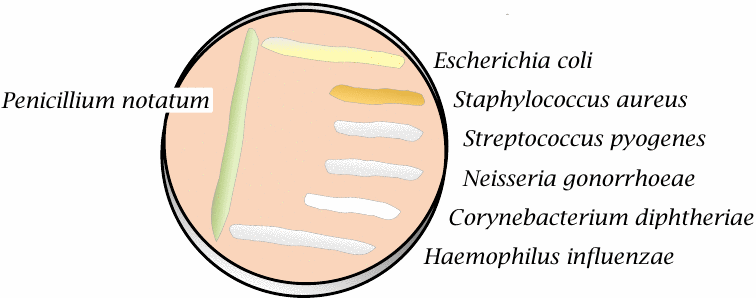
Some bacterial species are resistant to penicillin and therefore can grow right up to the mold colony. This is observed with Escherichia coli and Haemophilus influenzae, both of which are Gram-negative bacteria. Neisseria gonorrhoeae is Gram-negative also but is nevertheless inhibited by penicillin.
Most Gram-negative bacteria are resistant to penicillin because the drug cannot penetrate the outer membrane of these bacterial cells (see slide 11.4.1). In contrast, Gram-positive bacteria such as Staphylococcus aureus, Corynebacterium diphtheriae and Streptococcus pyogenes don’t have an outer membrane and are susceptible. Note, however, that these bacteria can still acquire resistance. S. aureus strains, in particular, have now mostly become resistant to penicillin. We will consider the underlying mechanisms of susceptibility and resistance to penicillin later (see slide 11.4.7ff.).
Chapter 15 covers the subject of drug discovery in a more detailed and systematic manner.
| 1.3.11 |
Drug development and approval |
- preclinical, in-house: synthesis, in vitro and preliminary animal testing
- investigational drug application to Food and Drug Administration (FDA)—must be approved before clinical testing
-
clinical trials in three phases:
- [(1)] Healthy volunteers; focus on pharmacokinetics, toxicity
- Small number of patients with targeted disease
- Larger patient collective (several hundred to several thousand), comparison to established reference therapies
- new drug application—review by FDA
- post-introduction market surveillance
Several of the historic examples may convey the impression that drug discovery can be done with modest means. That may have been true at the time; however, nowadays the process has become strongly regulated, and these regulations have turned drug discovery into an enormously laborious and costly exercise. The total cost of development of a new drug, from scratch, is on the order of one billion dollars. Therefore, while an academic setting may suffice to support the initial stages of development, only major pharmaceutical companies have the means to see the entire process through to the end.