| 8 |
Pharmacology of nitric oxide |
| 8.1 |
Physiological significance of nitric oxide (NO) |
- Powerful vasodilator—NO-releasing drugs are used in the treatment of cardiovascular disease
- Neurotransmitter—signaling in the CNS and the autonomic nervous system
- Inflammatory mediator—inhibition of NO synthesis is of interest as a therapeutic strategy in infection and chronic inflammation
Nitric oxide differs in many ways from other signaling molecules, and because of its unusual nature was discovered relatively late. Its first physiological function to be discovered was the relaxation of blood vessels. While this function remains the focus of its application in pharmacotherapy, a range of other effects with at least potential applications in clinical practice were characterized subsequently.
The three listed physiological functions correspond to three different isoforms of nitric oxide synthase, the enzyme that produces NO (see slide 8.3).
| 8.2 |
Identification of “endothelium-derived relaxing factor” as nitric oxide |
Vasorelaxation mediated by nitric oxide is controlled by the autonomic nervous system. The physiological experiments that uncovered the role of the endothelium, and subsequently of NO, in vasorelaxation were simple yet ingenious. In order to appreciate them, we need a little more anatomy.
| 8.2.1 |
Cholinergic and adrenergic nerve endings in a blood vessel wall |
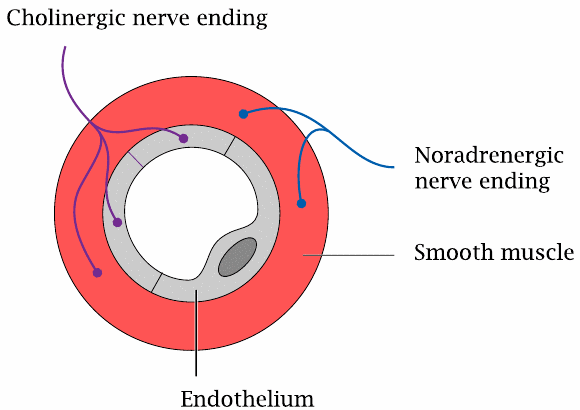
We had seen earlier that both the sympathetic and the parasympathetic nervous system innervate blood vessel walls and regulate their contraction and wall tension (slide 6.13.1). The adrenergic nerve endings of the sympathetic nerve fibers are found in the muscular layer of the vessel walls. In contrast, cholinergic nerve endings are found both in the muscular and the endothelial layers.
The wall tension of the blood vessel is sustained by the muscular layer, not the endothelium; therefore, the cholinergic innervation of the endothelium may be surprising. As it turns out, however, it is needed for the vasorelaxation and vasodilation induced by the parasympathetic system.
| 8.2.2 |
The endothelium is required for vascular relaxation in response to acetylcholine |
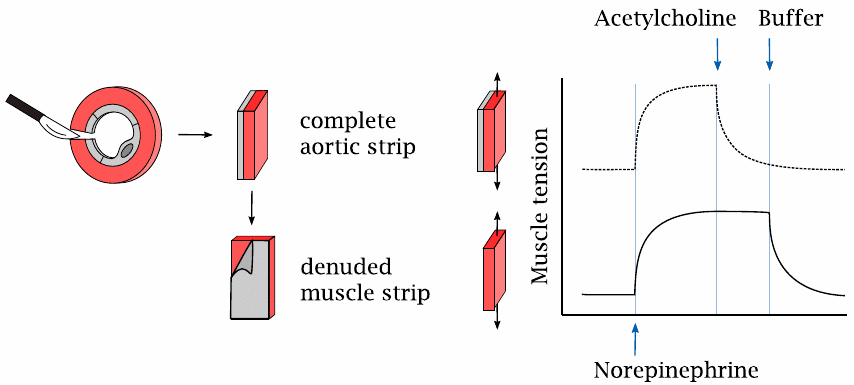
This slide illustrates the experiment that led to the discovery of nitric oxide-mediated vasorelaxation.
Contraction and relaxation of the vascular smooth muscle cells in response to norepinephrine and acetylcholine were studied with aortic strips. In this technique, slices are cut from an animal’s aorta and then opened with a radial incision. The endothelium can be left in place or peeled away in order to study its effect on the muscular layer. The resulting intact or denuded aortic strips were mounted between two distending hooks to measure their contractile force.
Application of norepinephrine induced contraction in strips with or without attached endothelium, as would be expected from the innervation pattern of the sympathetic nerve fibers. In contrast, norepinephrine-induced contraction could be reversed by acetylcholine only if the strip retained the endothelium (top) but not when the endothelium had been peeled away.
Interestingly, relaxation of a denuded muscular strip could be restored if the endothelial side of an intact strip was strapped onto it (not illustrated). These observations showed that, in response to cholinergic stimulation, the endothelium releases a diffusible substance that enters the muscular layer and induces its relaxation. This “endothelium-derived relaxing factor” was subsequently isolated and identified as NO.
| 8.3 |
The nitric oxide synthase reaction |
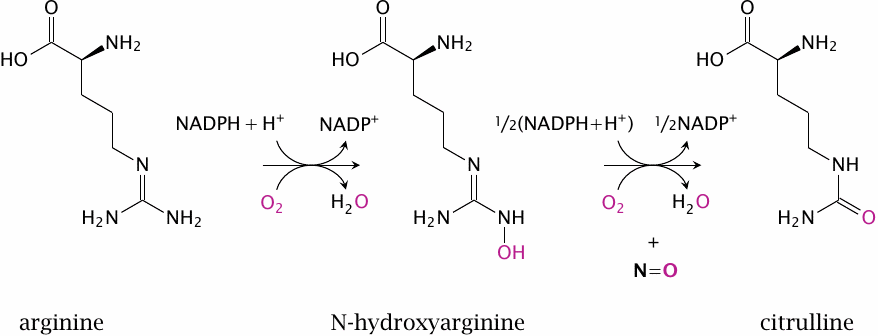
Nitric oxide is synthesized intracellularly by nitric oxide synthase (NOS). This reaction is rather complex and involves two successive monooxygenase steps. In the first step, arginine is converted to N-hydroxyarginine (NOHA), which is cleaved in the second step to NO and citrulline.
NOS occurs in several variations. Endothelial NOS (eNOS) and neuronal NOS (nNOS) are found in the eponymous cell types. Inducible NOS (iNOS) is found mainly in inflammatory cells. All three enzymes are homologous and perform the same reaction, but they differ in their regulatory properties.
| 8.3.1 |
Activation of NOS by calcium and calmodulin |
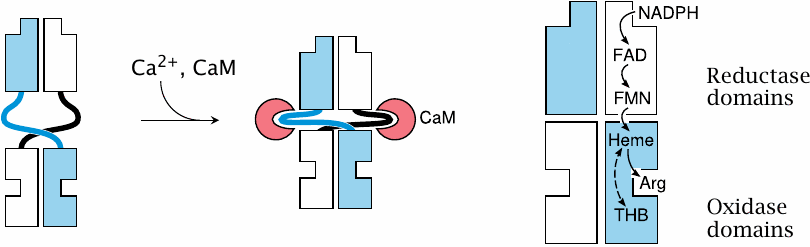
Nitric oxide synthase exists as a dimer. Each subunit contains a reductase domain and an oxidase domain. The reductase domain accepts electrons from NADPH and hands them over to the oxidase domain of the other subunit.
The reductase and oxidase domains are loosely connected by flexible hinges that, when extended, prevent the flow of electrons between them. The two domains are brought into close, productive interaction when calmodulin (CaM) binds to the hinges and changes their conformations. In the case of eNOS and nNOS, CaM binding only happens if the cytosolic level of Ca++ is elevated, which occurs only transiently when triggered by action potentials or through activation of GPCRs, such as for example the muscarinic ones in endothelial cells.
In contrast, inducible NOS binds calmodulin very avidly regardless of the calcium concentration, and therefore is active even at the low resting level of Ca++. Its activity is regulated not by short-term calcium signals but instead by transcriptional induction.
| 8.4 |
NO activates soluble guanylate cyclase (sGC) |
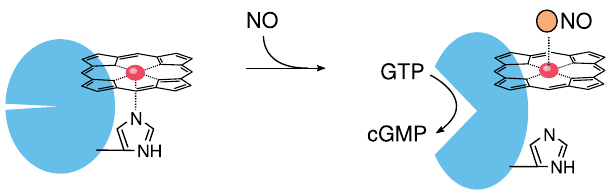
Between the last slide and this one, we have skipped an important step, namely the diffusion of NO out of the cell of origin, for example an endothelial cell, and into another one, such as a vascular smooth muscle cell. Like other small gas molecules, nitric oxide passes through cell membranes with ease, which enables it to directly and rapidly interact with intracellular targets.
Within its target cell, NO binds to the heme group of soluble guanylate cyclase (sGC), dislodging a strategic histidine residue. This causes a conformational change that activates the enzyme, which then begins to make cyclic GMP (cGMP). The production of cGMP as a second messenger is the most important signal downstream of NO.
| 8.4.1 |
Signaling effects of cGMP |
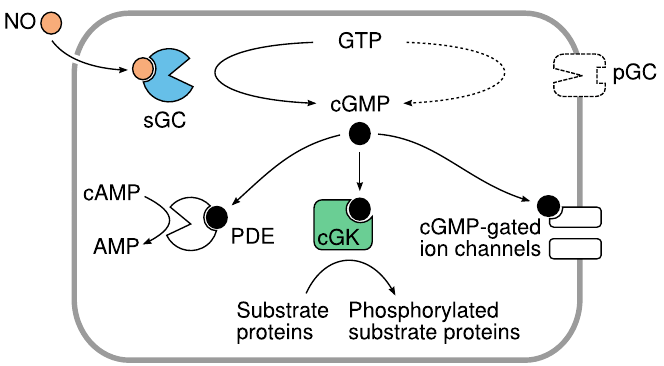
Like cAMP, cGMP targets multiple effector molecules inside the cell. The activation of cGMP-dependent protein kinases (cGK) is the most important single mechanism. Phosphodiesterase 5 (PDE) is activated, too, reducing the levels of both cAMP and cGMP. Actuation of cyclic nucleotide-gated cation channels affects the membrane potential and the cellular calcium level.
Membrane-bound receptor or “particulate” guanylate cyclases (pGC) provide an alternate means of cGMP production that is independent of NO but instead is controlled by several peptide hormones.
| 8.4.2 |
NO-induced relaxation of smooth muscle cells is mediated by cGK |
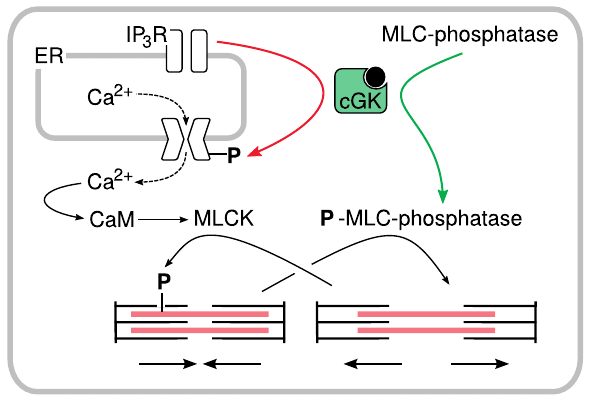
The relaxation of smooth muscle that occurs downstream of endothelial NO release is mediated by cGMP-dependent protein kinase. This kinase phosphorylates and thereby activates myosin light chain phosphatase. The phosphatase dephosphorylates myosin, which interrupts interaction of myosin with actin and induces relaxation.
cGK also phosporylates a regulatory subunit of the IP3 receptor channel. As we have seen before (slide 5.3.2), this channel mediates the outflow of Ca++ from the ER to the cytosol. Phosphorylation of the IP3 channel reduces Ca++ flow and therefore the calmodulin-dependent activation of myosin light chain kinase (MLCK).
It is worth noting that the effects of NO cut in below the GPCRs that mediate the effects of major vasoconstrictors such as angiotensin and norepinephrine. Therefore, NO-releasing drugs offer a means to interrupt the out-of-control signaling by such mediators that occurs in hemodynamic shock or hypertensive crisis.
| 8.5 |
S-nitrosylation of cysteine residues in proteins by NO |
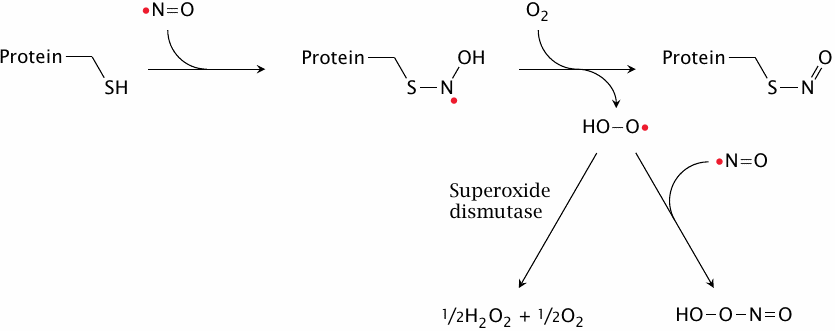
In addition to the cGMP-mediated signaling effects, nitric oxide can also influence protein function through reacting covalently with them. The reaction involves single cysteine residues in proteins, which become S-nitrosylated. The reaction also requires molecular oxygen, which is converted to superoxide. While the latter has been observed in vitro to react with a second molecule of NO to form peroxynitrite, it seems likely that in vivo most superoxide would be scavenged by superoxide dismutase.65
One might expect this modification to be too indiscriminate to be of use in selective and directed control of cell physiology. However, it has been demonstrated experimentally that S-nitrosylation is indeed quite selective in vivo.
| 8.5.1 |
Transfer of nitrosyl groups between proteins by glutathione |
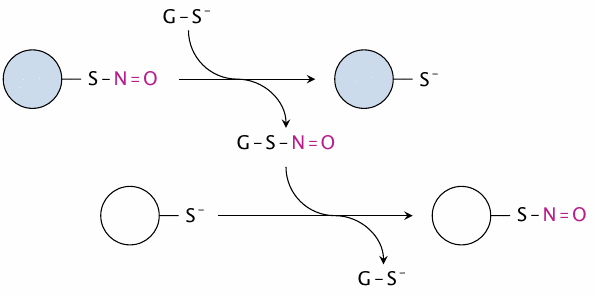
One mechanism that may contribute to the selectivity of protein-S-nitrosylation in vivo is the transfer of nitrosyl groups between proteins, which is mediated by glutathione. In this way, the nitrosyl groups can travel around the cell before settling down on the proteins with the thermodynamically most favorable cysteine residues.
Since glutathione is the most abundant thiol in the cell, this also means that nitrosylated glutathione functions as a reservoir of S–N=O (nitrosothiol) groups in the cell. In fact, many in vitro studies on the regulatory effects of protein-S-nitrosylation have used nitrosylated glutathione as the source of nitrosyl group instead of free NO for simplicity.
| 8.5.2 |
Identification of cysteines affected by S-nitrosylation: The biotin switch method |
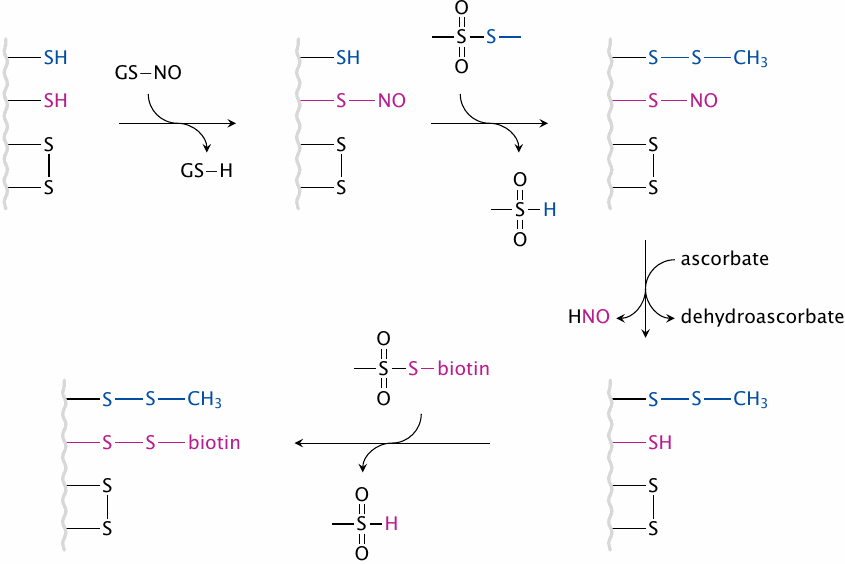
To understand the regulatory effects of protein-S-nitrosylation, it is important to identify the proteins, and the specific cysteine residues within them, that actually become S-nitrosylated in vivo upon NOS activation. The biotin switch method is an experimental protocol that permits the identification of these cysteines by selectively attaching biotin to them. This procedure involves the following steps:
- 1.After NOS has been activated and protein-S-nitrosylation has run its course, proteins are extracted from cells. Their remaining free SH groups are converted to disulfides using S-methyl-methanethiosulfonate.
- 2.Ascorbic acid is used to selectively reduce S–NO groups, while leaving native or synthetic disulfide bonds intact.
- 3.The reduced SH groups are labeled with a biotinylation reagent, via disulfide formation (as shown here) or other coupling chemistries.
After this procedure, biotin will be attached specifically to those cysteine residues that had been nitrosylated before. The biotinylated proteins can then be purified or selectively detected on a blot using the very strong and specific interaction between biotin and streptavidin.
| 8.5.3 |
Identification of proteins subject to nNOS-dependent S-nitrosylation |
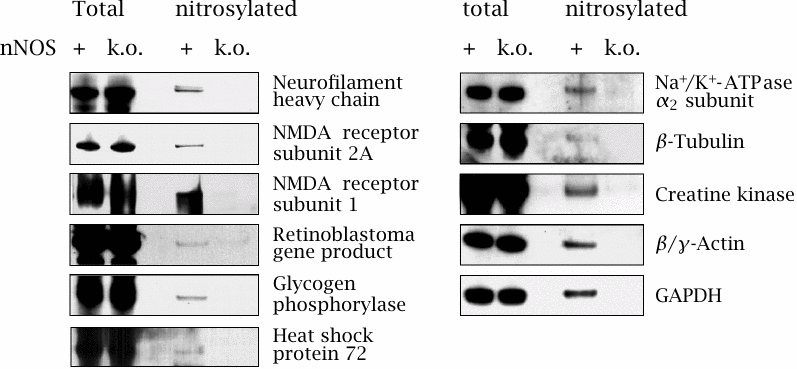
This experiment illustrates the selectivity of protein-S-nitrosylation in brain tissue from mice due to activation of neuronal NOS. The role of nNOS was ascertained by comparing the extent of nitrosylation between wild-type and nNOS k.o. mice.
After extracting the proteins from homogenized tissue, S-nitrosyl groups were converted to biotin derivatives as outlined above, and selectively detected on blots using labeled streptavidin. The total amount of each protein—nitrosylated or not—was imaged with antibodies and seems indistinguishable between wild-type and nNOS k.o. mice in every case. With each protein, the extent of S-nitrosylation is represented by the intensity of the third sample relative to the first one.
In all cases, S-nitrosylation is caused by nNOS, as shown by its absence in the knockout mice. The extent of nitrosylation is particularly strong with the NMDA receptor, and indeed this receptor—one of the ligand-gated glutamate receptors (see section 6.11)—is known to be inhibited by S-nitrosylation. Glyceraldehyde-3-dehydrogenase (GAPDH) is also quite strongly affected. Intriguingly, this protein does not only serve in its catalytic role in glycolysis but moonlights as a signaling molecule in the control of apoptosis (programmed cell death). Figure adapted from [68].
| 8.6 |
Role of NO and iNOS in the killing of microbes by phagocytes |
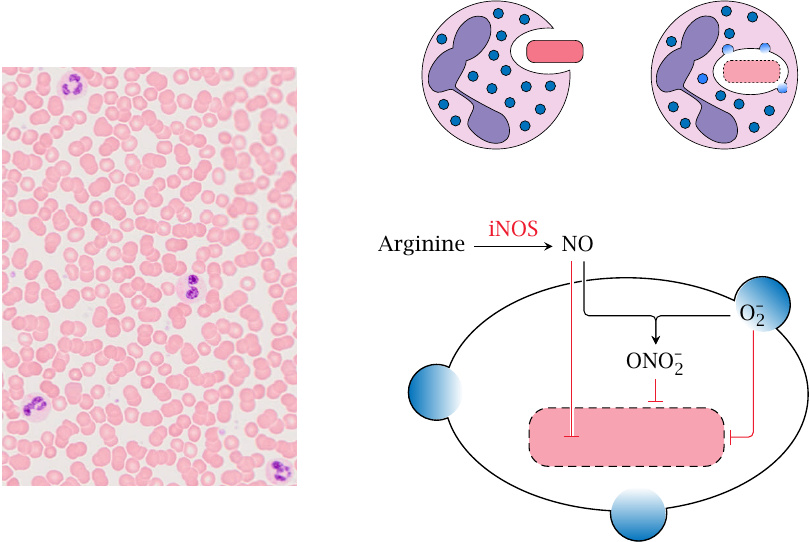
NO released by eNOS and nNOS serves signaling roles and is subject to rapid, calcium-dependent regulation. In contrast, NO produced in cells of the immune system primarily serves as an inflammatory effector. Its production doesn’t need to occur as rapidly but must be sustained for longer, and accordingly it is regulated by transcriptional induction of the enzyme rather than calcium and calmodulin. This form of the enzyme is therefore called inducible NOS (iNOS).
The antimicrobial effect of NO is strongest in combination with reactive oxygen species. In phagocytosis, microbes are ingested by granulocytes or macrophages and then end up inside intracellular vesicles called phagosomes. Peroxisomes then fuse to the phagosomes, releasing O2•− (superoxide; produced by NADPH oxidase) and H2O2 (produced by superoxide dismutase). Concomitantly, iNOS is activated in the cytosol. NO enters the phagosomes, where it may combine with superoxide to form peroxynitrite (ONOO−), which is strongly microbicidal. Alternatively, NO itself may diffuse across the microbial cell wall to wreak havoc inside.66
As with reactive oxygen species, the production of NO by inflammatory cells is a mixed blessing—important in antimicrobial defense, but destructive in rheumatic or other autoimmune disease. In severe, systemic infection (septicemia), the massive release of NO by immune cells causes relaxation of the vasculature, leading to a dangerous drop in blood pressure (septic shock). Therefore, pharmacological inhibition of iNOS is of great medical interest.
| 8.7 |
NO-releasing drugs |
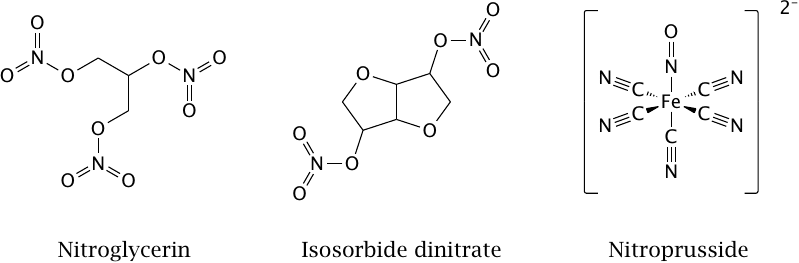
The drugs shown in this slide are used in the treatment of hypertension and of stenotic coronary artery disease. The first drug to be so employed was nitroglycerin, which is more commonly known as an explosive. Its therapeutic properties were discovered when workers exposed to nitroglycerin fumes at Mr. Alfred Nobel’s factory complained about headaches recurring at the beginning of each work week (headaches are often caused by dilating blood vessels in the skull).
Nitric oxide is released from nitroglycerin by various enzymes. The drug is promptly absorbed across mucous membranes, and sublingual application of spray or droplets is used by patients with acute attacks of angina pectoris. Isosorbide dinitrate is used by the same group of patients, but has a slower and longer lasting effect than nitroglycerin and is used orally for sustained therapy.
Sodium nitroprusside releases NO spontaneously, without enzymatic catalysis. It is the most powerful NO-releasing drug and is used, by intravenous infusion, in hypertensive crisis and other emergencies involving dangerously high blood pressure. Somewhat counterintuitively, it is also used in hemodynamic shock, which involves extremely low blood pressure. In this condition, perfusion of the kidneys and other interior organs is reduced by autonomic reflexes to a dangerous extent. After stabilizing blood pressure with other means, nitroprusside is used to break through this reflex blockade and restore perfusion to the endangered organs.
In addition to NO, nitroprusside also releases cyanide. This is potentially toxic but can be neutralized by the simultaneous application of sodium thiosulfate, which reacts with cyanide to form thiocyanate.
| 8.7.1 |
Bioactivation of nitroglycerin by mitochondrial aldehyde dehydrogenase |
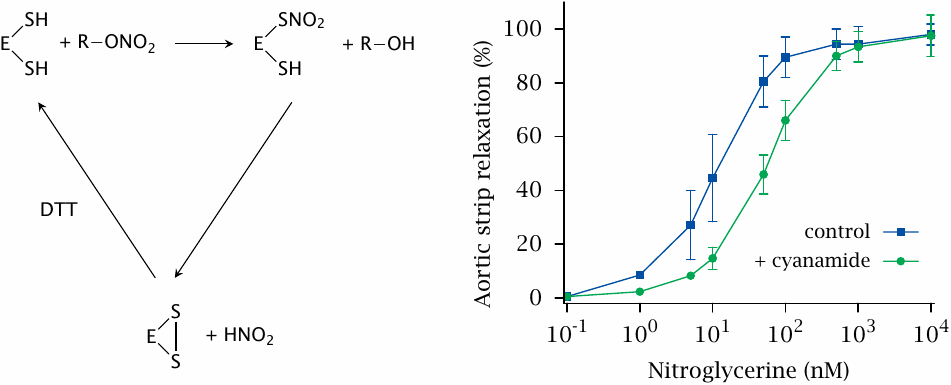
Nitroglycerin and isosorbide dinitrate can be activated by multiple enzymes. In mice and several other species, an important enzyme is mitochondrial aldehyde dehydrogenase (ALDH). This slide shows the reaction mechanism—in vitro, the reaction requires the addition of a thiol-reducing agent such as dithiothreitol (DTT), and the reaction produces S-nitrosylated intermediates—as well as the effect of inhibiting ALDH with cyanamide on the relaxation of aortic strips ([69]; compare slide 8.2.2). Genetic knock-out of ALDH suggests an even greater contribution of ALDH to nitroglycerine bioactivation; however, this varies with the drug and the animal species in question.
| 8.7.2 |
Bioactivation of nitroglycerin and ISDN by human cytochrome P450 isoforms |
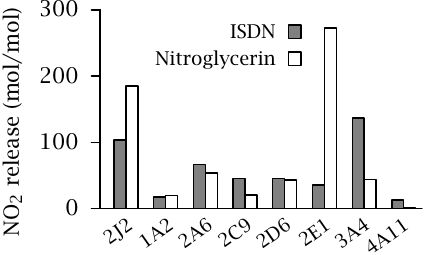
While both nitroglycerin and isosorbide dinitrate are metabolized by cytochrome P450 enzymes, a more detailed analysis shows that the enzyme isoforms with the strongest activity toward each are somewhat different: CYP2E1 is highly active with nitroglycerin, whereas CYP3A4 is more active with ISDN. Both drugs are converted efficiently by CYP2J2, which is strongly expressed in blood vessels and is likely important in the prompt clinical effect of nitroglycerin.
As we have seen, cytochrome P450 enzymes contain heme. Like the heme in sGC, the one in cytochrome P450 may bind NO; this will inactivate the enzyme. Inactivation of cytochrome P450—and/or aldehyde dehydrogenase, which is subject to protein-S-nitrosylation—by NO released from drugs contributes to nitrate tolerance, which develops upon prolonged drug application and can only be broken by pausing the use of these drugs.67
In the experiment shown, the CYP isoforms were recombinantly expressed in yeast. Experimental systems such as this one are often used to study the susceptibility of novel drug molecules to metabolism during preclinical development. Figure prepared from original data in [70].
| 8.7.3 |
NO release by molsidomine |
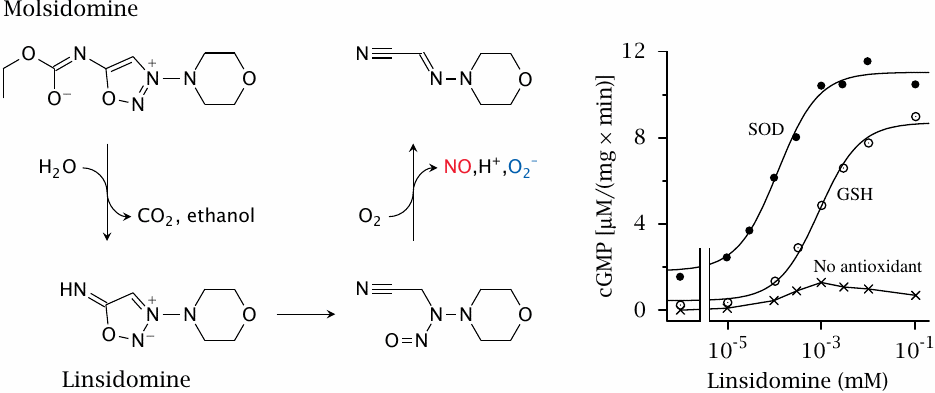
Molsidomine is an alternative NO-releasing drug that does not require metabolism by cytochrome P450 or aldehyde dehydrogenase, and therefore is less prone to the development of tolerance. The left panel shows the mechanism of NO release: Enzymatic hydrolysis of molsidomine yields linsidomine, which then decomposes spontaneously. The final step depends on O2 and yields both NO and superoxide.
As we had seen above (slide 8.6), superoxide can combine with NO to form peroxynitrite. Accordingly, in order to render the NO released by linsidomine available for the activation of guanylate cyclase, superoxide must be scavenged. This is illustrated in the experiment in the right panel, which shows that appreciable amounts of cGMP are formed downstream of linsidomine decay only in the presence of scavenging agents such as superoxide dismutase (SOD) or glutathione (GSH). Figure prepared from original data in [71].
| 8.8 |
Antiinflammatory effect of iNOS inhibition |
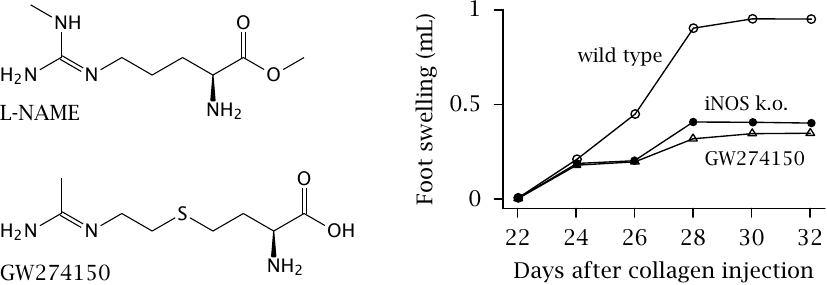
The activity of iNOS in inflammation is potentially destructive and contributes to tissue damage in rheumatism and other autoimmune diseases; therefore, inhibiting iNOS is potentially useful. To make this work, it is important to avoid inhibition of eNOS and nNOS. While most available NOS inhibitors, such as l-N-nitroarginine-methyl ester (l-NAME) inhibit all NOS isoforms, some are selective for iNOS. One such drug the experimental inhibitor GW274150.
In the experiment on the right, the effectiveness of GW274150 was studied in a mouse model of inflammation. Collagen-induced arthritis can be provoked in mice by injecting their paws with bovine collagen type II, to which the mice then mount an immune reaction that leads to swelling and inflammation.
After injection on day 0, the mice take three weeks to produce antibodies and then develop inflammation. The intensity of this inflammation can be measured by the extent of paw swelling. Genetic knock-out of iNOS and the experimental drug are equally effective in mitigating inflammation. Figure prepared from original data in [72].
| 8.8.1 |
Structure and mode of action of sildenafil (Viagra™) |
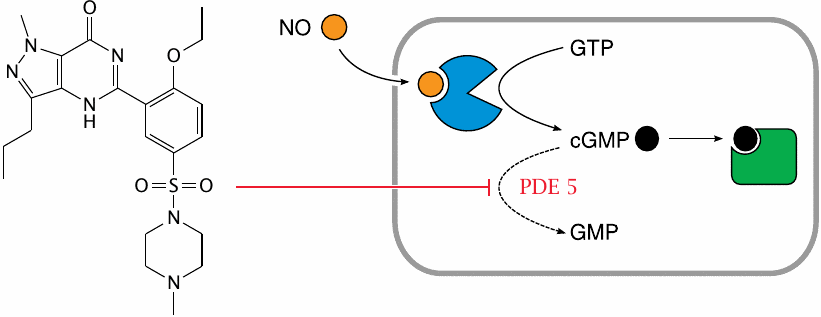
While NO-releasing drugs influence the contractile activity of smooth muscle cells by increasing cGMP, an equally viable approach should be the inhibition of cGMP degradation by phosphodiesterase. This rationale led to the development of sildenafil, which was originally intended as yet another cardiovascular drug for the elderly.
As it turned out, the drug proved very popular with the old folks alright, but not entirely for the intended reasons. Nevertheless, in addition to its well-known effect on male potency, the drug also does promote vasorelaxation, which can prove dangerous when used in combination NO-releasing drugs or similar.
Prompted by the risks of sildenafil use in cardiac patients engaging in strenuous exercise, some administrative districts (Kantons) in Switzerland have mandated the training of prostitutes in the use of defibrillators.