| 7 |
Hormones |
| 7.1 |
Major types of hormone receptors |
-
G protein-coupled receptors
- Many peptide hormones: glucagon, hypothalamic and hypophyseal hormones
- Epinephrine, norepinephrine
-
Receptor tyrosine kinases
- Insulin
- Growth hormone and growth factors
-
Nuclear hormone receptors
- Steroid hormones
- Thyroid hormones
Hormones are fundamental to metabolic and other physiological regulation. Accordingly, hormone receptors, as well as enzymes involved in hormone synthesis, are important drug targets.
Hormone receptors fall into different functional classes. We have already encountered numerous GPCRs, and we have met some nuclear hormone receptors in the context of drug metabolism, namely the pregnane X receptor and the retinoid X receptor.
The receptor tyrosine kinases (RTKs) are another major receptor class. Like GPCRs, these receptors sit in the cytoplasmic membrane, bind their ligands on the extracellular side, and transmit the signal to the cell interior through a conformational change. However, unlike GPCRs, the RTKs do not transmit their signal through noncovalent interaction with downstream adapters. Instead, the activated receptors themselves function as protein kinases. RTKs typically phosphorylate multiple substrate proteins, which then noncovalently bind and activate various downstream regulatory proteins.
Important examples of the RTK class are the receptors for insulin and for growth hormone. Several other receptor tyrosine kinases are involved in tumor initiation and growth. We will learn a bit more about these in Chapter 12.
| 7.2 |
The hypothalamic-pituitary axis |
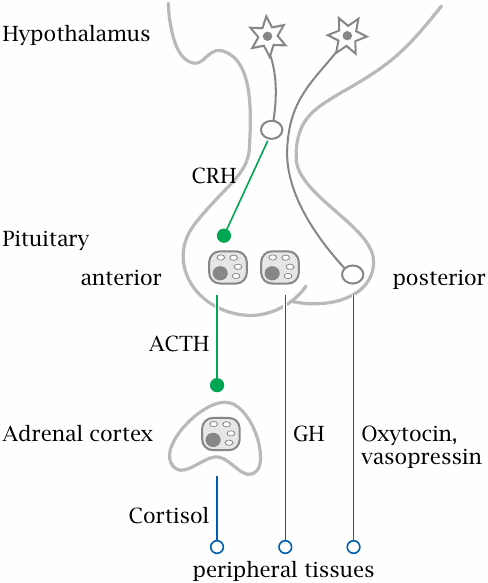
Hormone-secreting glands are also referred to as endocrine glands; this name reflects the fact that secretion occurs into the body interior, that is, into the bloodstream. In contrast, exocrine glands are the ones that secrete their products across the skin or the mucous membranes.
Many of the endocrine glands are under control by the hypophyseal gland or pituitary, which in turn is controlled by the hypothalamus. These two are located next to each other at the base of the brain. The hypothalamus is itself part of the brain, whereas the hypophyseal gland extends downward from it. Hypothalamic neurons extend their axon endings into the hypophyseal gland, which peeks out of the blood brain barrier and is able to release hormones into the systemic circulation; one could say that the hypophyseal gland is an outlet for the brain through which it releases hormones to the body.
The hypophyseal gland consists of an anterior and a posterior lobe. The posterior lobe contains just the axon endings of hypothalamic nerve cells, which secrete two related peptide hormones, oxytocin and vasopressin, directly into the circulation.
The anterior pituitary lobe contains gland cells that are controlled by specific hypothalamic peptide hormones, for example corticotropin releasing hormone (CRH). In response to these hypothalamic hormones, the cells of the anterior pituitary release peptide hormones that stimulate peripheral glands. One of these is adrenocorticotropic hormone (ACTH), which stimulates the cortex of the adrenal glands; another one is thyroid-stimulating hormone (TSH), which controls the thyroid gland. In addition to hormones that control peripheral endocrine glands, the anterior pituitary also produces prolactin, which stimulates the mammary glands, as well as growth hormone (GH).55
| 7.2.1 |
Peripheral glands and hormones controlled by the anterior pituitary |
| Gland | Stimulated by | Hormones produced |
| adrenal glands (cortex) | adrenocorticotropic hormone (ACTH) | gluco-, mineralocorticoids; androgens |
| thyroid gland | thyroid-stimulating hormone (TSH) | tri-, tetraiodothyronine (T3, T4) |
| testicles / ovaries | follicle-stimulating hormone (FSH), luteinizing hormone (LH) | androgens, estrogens, progestins |
| diffuse | growth hormone | growth factors |
| mammary gland | prolactin | — |
| 7.2.2 |
Regulatory patterns in the hypothalamic-pituitary axis |
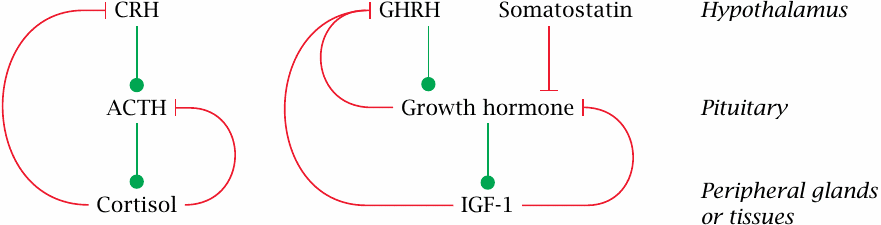
This scheme illustrates two different regulatory patterns that occur in the hypothalamic-pituitary axis. The production of cortisol by the adrenal glands is stimulated by adrenocorticotropic hormone (ACTH), which in turn is stimulated by hypothalamic corticotropin-releasing hormone (CRH). Cortisol exercises negative feedback on both. The control of the thyroid gland by thyroid-stimulating hormone (TSH) and thyrotropin-releasing hormone (TRH) works analogously, as does the regulation of sexual hormone secretion by endocrine cells in the ovaries and testicles (see later).
The secretion of growth hormone (GH) by the anterior pituitary is controlled by two hypothalamic hormones; it is activated by growth hormone releasing hormone (GHRH) and inhibited by somatostatin. GH itself stimulates the production of insulin-like growth factor 1 (IGF-1) in the liver and other tissues. Both GH itself and IGF-1 exert negative feedback on GH secretion.
Keep in mind that these homeostatic feedback loops are only part of the story. Spontaneous variations in hormone levels, such as the early-morning high of cortisol, the spike of growth hormone secretion shortly after onset of sleep, or the variations of estrogens and progestins that occur during the menstrual cycle, involve additional control mechanisms. This is where the brain comes in—for simple homeostatic feedback alone, we wouldn’t need the brain; peripheral glands such as the parathyroids are perfectly capable of it (see section 7.5).
| 7.2.3 |
The posterior lobe of the hypophyseal gland produces oxytocin and vasopressin |
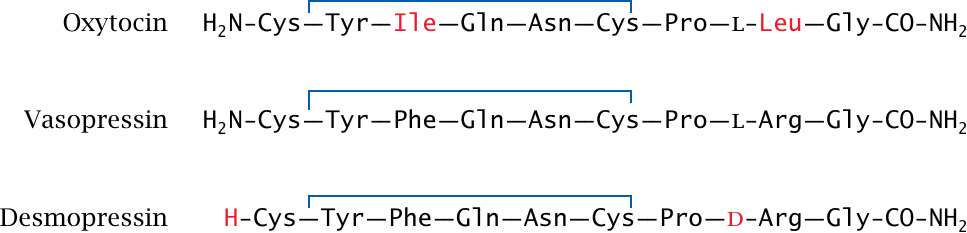
Both oxytocin and vasopressin are derived from precursor polypeptides; the amido groups at their C-termini are derived from a glycine residue that gets oxidatively truncated after proteolytic cleavage of the precursor. Each of the two hormones acts on cognate GPCRs that couple prominently to the phospholipase C cascade (see slide 5.3.2).
Oxytocin drives the contraction of smooth muscle in the uterus during labor; it is also used as a drug to induce labor when its spontaneous onset is delayed.56 Moreover, it is released during breast feeding and induces contraction of smooth muscle in the milk ducts of the mammary glands, which assists in the release of the milk.
Vasopressin, also known as antidiuretic hormone, activates smooth muscle in the circulation; it also acts on cells in the distal tubules of the kidney, where it promotes the preservation of water. It is used as a drug in individuals with deficient endogenous production, a fairly rare condition that is known as diabetes insipidus.
Desmopressin is a synthetic vasopressin analogue in which the N-terminal amino group has been removed, and the l-arginine residue has been replaced by the d-enantiomer. Both of these modifications slow the degradation of the molecule by circulating peptidases. Like the native hormone, it can be in the treatment of diabetes insipidus.
| 7.3 |
Thyroid hormones |
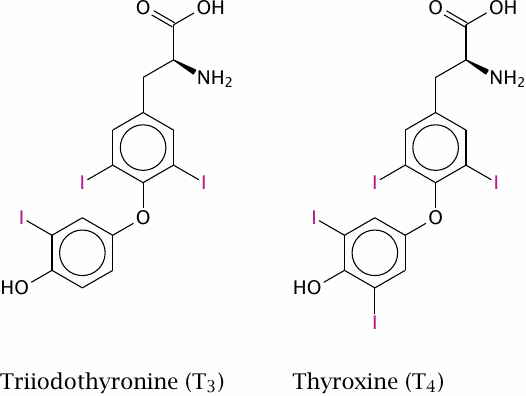
The thyroid hormones triiodothyronine (T3) and tetraiodothyronine (thyroxine, T4) are produced by the thyroid gland, which is under the control of the thyroid-stimulating hormone from the anterior pituitary. These hormones have broad and diverse activating effects on human physiology, which are mediated through transcriptional regulation. For example, they increase the expression of β-adrenergic receptors, boosting heart rate and ejection volume, and of respiratory chain-uncoupling proteins, which cause metabolic substrates to be burned without ATP production.57
Lack of thyroid hormones in children due to defective thyroid gland development causes delayed and stunted physical and mental development. If diagnosed in time, this can be corrected by hormone substitution treatment.
| 7.3.1 |
Thyroid hormones activate cognate nuclear hormone receptors |
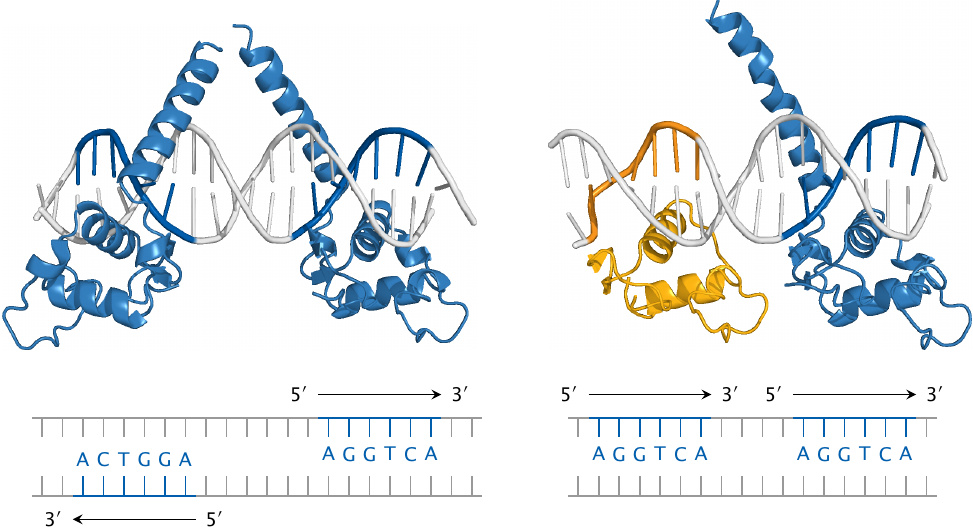
The thyroid hormones activate cognate nuclear hormone receptors; these are similar to the pregnane X receptor (see slide 4.3) but bind to different recognition elements. The thyroid hormone receptors come in α and β versions, and further diversification arises from the formation of various receptor homo- or heterodimers, which then bind to different recognition sequences on the DNA.
This slide illustrates the recognition of specific DNA sequences (thyroid hormone response elements) by thyroid receptor (TRβ, blue) and retinoid X receptor (RXRα, yellow). Both monomeric receptors recognize the same sequence element, namely, the hexanucleotide AGGTCA. However, different receptor dimers recognize different target sequences nevertheless; this is due to variations in the relative orientations of the DNA-binding domains within these dimers.
Left: The TRβ homodimer recognizes two instances of the consensus sequence that occur on opposite strands and point away from one another; such sequence motifs are referred to as inverted palindromes.
Right: The TRβ–RXRα heterodimer recognizes two instances of the consensus sequence that are located on the same strand (direct repeats). In both panels, only the DNA-binding domains of the receptor molecules are shown; the remaining domains would attach to the tips of the upward pointing helices.
| 7.3.2 |
Tissue structure of the thyroid gland, and localization of hormone synthesis |
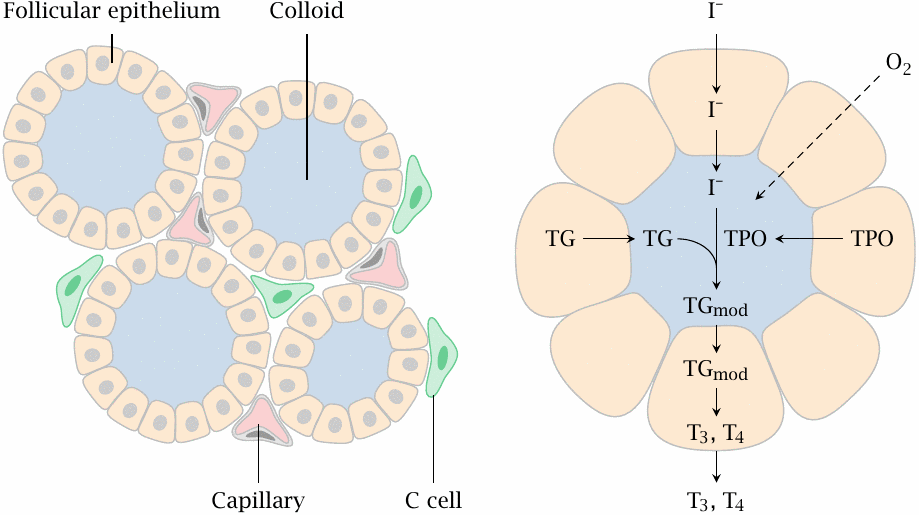
The thyroid gland has a peculiar tissue structure that is related to its likewise unusual biochemical function. The crucial and characteristic steps in thyroid hormone synthesis occur in confined extracellular compartments, little “reactors”, which are referred to as thyroid follicles.
Left: Follicular epithelial cells enclose the follicle lumen, which is filled with protein-rich colloid. C-cells are located outside the follicles; they are unrelated to T3 and T4 synthesis but instead produce the peptide hormone calcitonin (see slide 7.5.2).
Right: The cells of the follicular epithelium cells synthesize the proteins thyroglobulin (TG) and thyroid peroxidase (TPO) and secrete them into the follicle lumen. They also take up iodide and transport it into the follicle lumen, where it is used by thyroid peroxidase for the iodination of tyrosine side chains of thyroglobulin. Thyroid peroxidase also catalyzes the subsequent step, in which it joins two iodinated tyrosine residues (see slide 7.3.4).
The peroxidase-modified thyroglobulin (TGmod) is taken up into the follicle cells again via endocytosis and proteolytically degraded, which yields the free hormones triiodothyronine (T3) and thyroxine (T4).
| 7.3.3 |
Tyrosine side chain iodination by thyroid peroxidase |
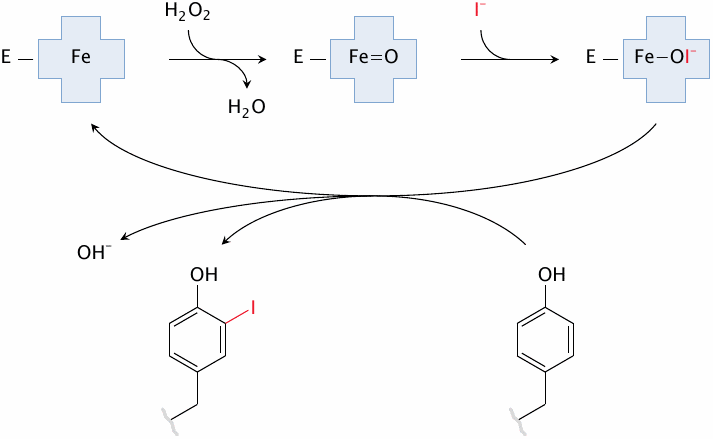
In the biosynthesis of the thyroid hormones, the first step is the iodination of tyrosyl residues in thyroglobulin. Thyroid peroxidase contains a heme moiety that generates hypoiodite from iodide and H2O2. The hypoiodite then reacts with the hydroxyphenyl side chain of tyrosine.
The slide shows the formation of monoiodotyrosine; diiodotyrosine is formed from monoiodotyrosine in another round of the same reaction. Both mono- and diiodotyrosine can participate in the subsequent coupling reaction (see next slide).
| 7.3.4 |
Coupling of two iodinated tyrosine side chains |
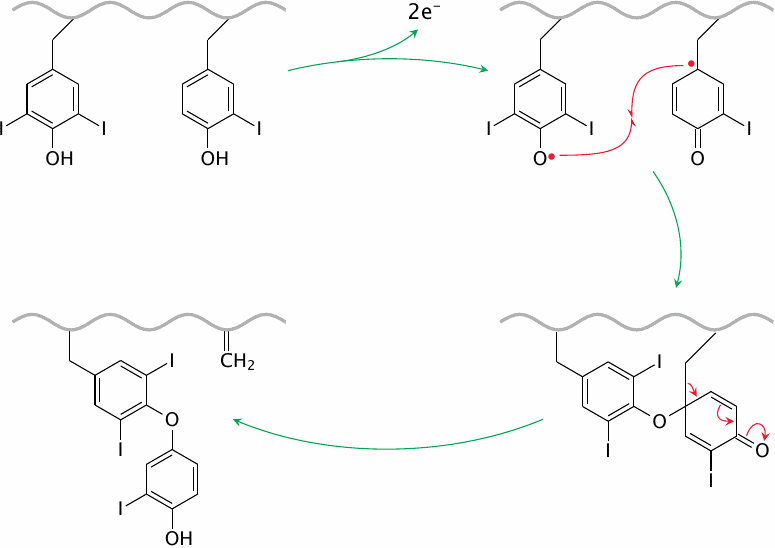
The coupling reaction between two iodinated tyrosyl side chains is also catalyzed by thyroid peroxidase. The enzyme abstracts an electron from each of the substrate side chains, which then undergo radical recombination. The reaction leaves a dehydroalanine residue in the protein backbone.
The slide shows the reaction of a diiodotyrosine residue with a monoiodotyrosine side chain; this will produce a precursor of T3. The same reaction can also occur between two diiodotyrosine side chains, which will produce a T4 precursor. After reuptake of the modified thyroglobulin by the follicular epithelial cells, T3 and T4 are liberated simply by proteolytic breakdown of the protein.
Both T3 and T4 are secreted into the bloodstream. Both are able to activate thyroid hormone receptors; however, T3 is much more active. T4 can be deiodinated to T3 and thus functions mostly as a reservoir of the latter. Drugs that inhibit the enzyme that carries out this deiodination step—the antiarrhythmic drug amiodarone (see slide 7.3.6) in particular—may cause a functional thyroid hormone deficit, although the combined serum levels of T3 and T4 may be normal.
| 7.3.5 |
Thyroid hormones and pharmacotherapy |
- Goiter: iodide supplementation
-
Hyperthyroidism due to anti-TSH-receptor autoantibodies or hormone-producing
tumors:
- thyroid peroxidase inhibitors
- radioiodine (131I)
- Lowering blood lipid levels: TR-β-selective agonists (KB-141)
- Interference with thyroid hormone release or conversion: lithium and amiodarone
The hypophyseal thyroid-stimulating hormone (TSH) stimulates the activity of existing gland cells, driving iodine accumulation and hormone synthesis. In addition, TSH also drives the growth of gland tissue. This dual effect is important for both pathogenesis and therapy.
A shortage of iodine will hamper thyroid hormone synthesis. The hypophyseal gland will produce more TSH, which will prompt excessive growth of the thyroid gland (goiter). The enlarged thyroid gland usually manages to supply enough thyroid hormones, so that symptoms other than goiter itself are usually absent. Goiter due to iodine deficiency used to be common but is now largely controlled by the iodide supplementation of cooking salt.
Both autoantibodies and tumors—benign or, less commonly, malignant—are common causes of hyperthyroidism. Autoantibodies directed at the TSH receptor, a GPCR, may activate it and trigger nonphysiological growth of the thyroid gland, as well as excessive thyroid hormone production. Increased metabolic turnover results in weight loss, accelerated heart rate, lowered blood lipids and other symptoms.
Treatment options for hyperthyroidism include inhibitors of thyroid peroxidase as well as surgery and radioactive iodide (131I−). The latter form of treatment takes advantage of the fact that only thyroid gland cells actively accumulate iodide and are therefore selectively destroyed. Importantly, in thyroid cancer, 131I− will also affect metastatic tumor cells, as long as these retain the ability to accumulate iodide. The accumulation of 131I− can be enhanced by simultaneous application of TSH.
Lithium inhibits the release of thyroid hormones from gland cells. While this can cause side effects when lithium is used to treat depression, it can be used to advantage in radioiodide treatment, since it will cause the hormone-incorporated 131I− to be retained longer inside the gland cells.58
| 7.3.6 |
Drugs that influence thyroid hormone function |
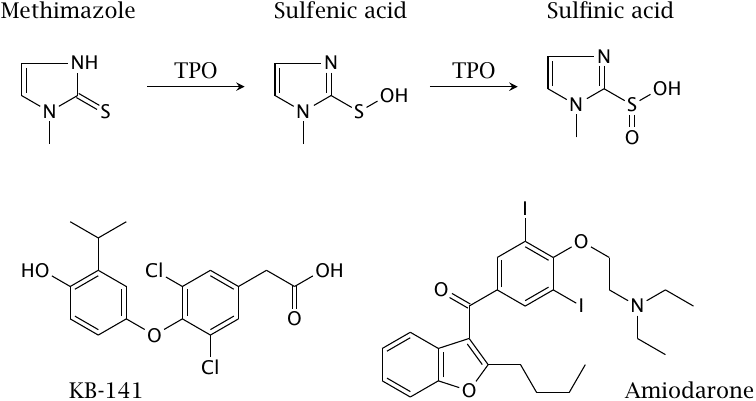
This slide shows some of the drugs whose modes of action were discussed above. Methimazole is a mechanism-based inhibitor of thyroid peroxidase that is used in hyperthyroidism. Shown next to methimazole are the putative oxidation products that are formed by thyroid peroxidase (TPO) and inactivate the enzyme by binding to its heme group.
KB-141 is a TRβ-selective thyroid receptor agonist. Such agonists lower blood lipid concentrations but avoid some other effects of nonselective receptor agonists; they are of interest in the treatment of hyperlipidemia. Amiodarone has been discussed in section 7.3.4.
| 7.4 |
Steroid hormones |
| Class | Major members | Glands |
| Glucocorticoids | Cortisol, cortisone | Adrenal glands |
| Mineralocorticoids | Aldosterone | Adrenal glands |
| Androgens | Testosterone, dihydrotestosterone | Testicles |
| Estrogens | Estradiol, estriol | Ovary |
| Progestins | Progesterone | Ovary, placenta |
Steroid hormones are all structurally related and biosynthetically derived from cholesterol. Glucocorticoids are involved in metabolic regulation, but their application as drugs is owed to their antiinflammatory and immunosuppressive activity. Mineralocorticoids regulate secretion and retention of sodium and potassium in the kidneys; this also has effects on blood pressure. Sexual steroids sustain the function of sexual organs and, in women, gestation.
Pharmacological applications exist for all functional classes of steroid hormones or related synthetic agonists and antagonists.
| 7.4.1 |
Effector mechanisms |
| Mechanism/target | Receptor binds to | Example |
| Transcriptional induction (transactivation) | DNA directly | Induction of enzymes of gluconeogenesis by cortisol |
| Transrepression | Other proteins that regulate transcription | Inhibition of transcription factors AP-1 and NF-κB by cortisol |
There is a cognate nuclear hormone receptor for each class of steroid hormones. All of these receptors act as transcription factors, much like the thyroid hormone receptors; this mode of action is referred to as transactivation. Glucocorticoids can also act via transrepression, which is mediated by direct protein-protein interaction between the ligand-bound glucocorticoid receptor and other transcriptional regulators.
Additional signaling mechanisms exist for some steroids. G protein-coupled receptors have been characterized that are activated by estrogens [59]. While conceptually intriguing, this has not yet resulted in pharmacological applications. So-called neurosteroids activate the GABAA receptor in the brain [60]; synthetic analogues such as alfaxalone can be used to induce narcosis.
| 7.4.2 |
Adrenal steroid hormones: functional classes |
| Class | Physiological function |
| Glucocorticoids | Metabolic regulation, strong anti-inflammatory action |
| Mineralocorticoids | Control of sodium and potassium elimination in the kidneys |
| Androgens | Precursors for gonadal androgen and estrogen synthesis |
The adrenal glands sit atop each of the two kidneys. They have two distinct zones, the outer cortex (“bark”) and the inner medulla (“marrow”). The steroid hormones are all synthesized in the cortex, while the medulla produces epinephrine and norepinephrine.
Glucocorticoids and mineralocorticoids are produced entirely in the adrenal gland and are released as finished products. In contrast, the adrenal androgens, which are formed in substantial amounts in the adrenal glands of both men and women, serve mostly as precursors for the gonadal synthesis of sexual hormones. Compared to the gonadal androgens testosterone and dihydrotestosterone, the adrenal androgens have low specific activity.
| 7.4.3 |
Synthesis of adrenal steroids |
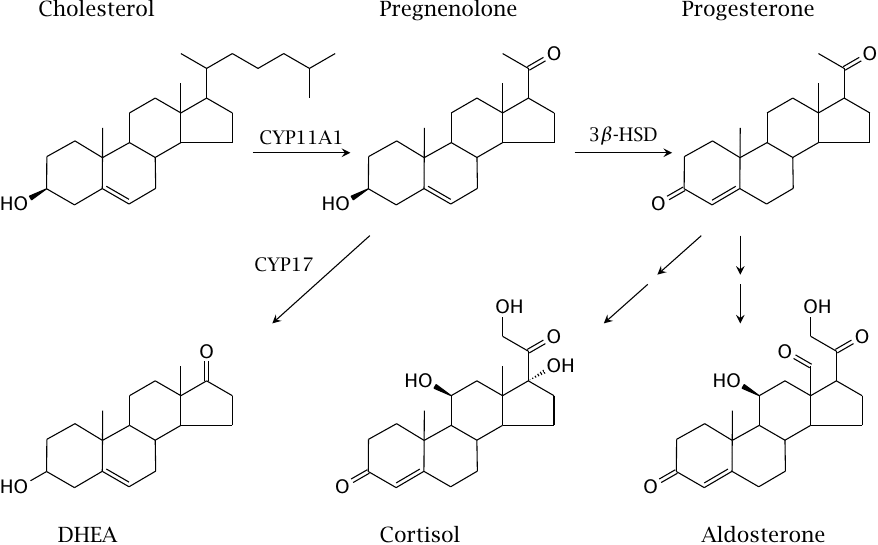
Aldosterone and cortisol are each derived from progesterone in three successive enzyme reactions; most of the enzymes involved are cytochrome P450 enzymes.
The key hormones released from the adrenal gland are cortisol, aldosterone, and dehydroepiandrosterone (DHEA), which are the major glucocorticoid, mineralocorticoid, and adrenal androgen, respectively. Progesterone is the most important progestin hormone; however, in the adrenal glands, it is not released to a significant extent but mostly serves as a synthetic intermediate, as does pregnenolone.
One of the steps in the synthetic pathway from progesterone to cortisol—the introduction of the hydroxyl group on the ring—is carried out by 11-β-hydroxylase (CYP11B). This enzyme also activates the anticancer drug mitotane, which therefore selectively targets tumors in the adrenal gland (see slide 12.4.3).
| 7.4.4 |
Glucocorticoids control inflammation via transrepression |
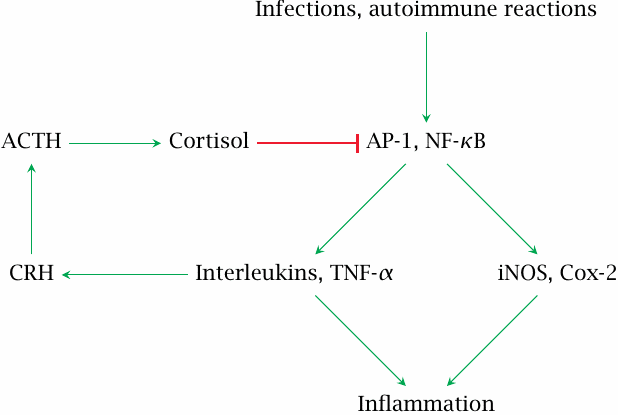
Cortisol and other glucocorticoids are powerful antiinflammatory agents, and most of their therapeutic applications are based on this activity. While direct transcriptional regulation participates, it seems that transrepression is responsible for much of the antiinflammatory activity.
The proinflammatory transcription factors AP-1 and NF-κB are active in inflamed tissue. They increase the expression of the pro-inflammatory enzymes cyclooxygenase 2 (Cox-2) and inducible nitric oxide synthase (iNOS),59 and also of inflammatory peptide mediators such as interleukins and tumor necrosis factor α, TNF-α.
Interleukins 1, 2 and 6 as well as TNFα stimulate the secretion of corticotropin-releasing hormone (CRH) in the hypothalamus. Cortisol released downstream of CRH activates the glucocorticoid receptor, which then inhibits AP-1 and NF-κB through transrepression. Glucocorticoids therefore are part of a negative feedback loop that keeps overall inflammatory activity in check.
| 7.4.5 |
Glucocorticoid receptor agonists and antagonists |
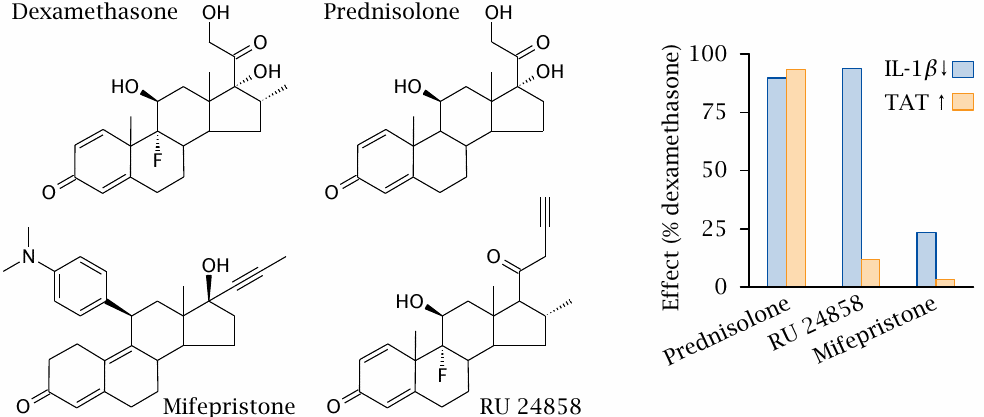
Dexamethasone and prednisolone are synthetic glucocorticoid receptor agonists that, like cortisol, exercise both antiinflammatory and metabolic effects. Mifepristone is a glucocorticoid receptor antagonist. In addition, it is also a progestin receptor antagonist; its more common medical applications relate to this latter activity.
In the experiment shown on the right (plot redrawn from [61]), the activities of these conventional drugs were compared to those of the experimental drug RU 24858. Downregulation of interleukin-1β (IL-1β) measures antiinflammatory activity and is mediated by transrepression. The induction of tyrosine transaminase (TAT) serves as a parameter of metabolic regulation via transcriptional induction. This enzyme participates in amino acid degradation and bolsters the supply of substrate carbon to gluconeogenesis. With dexamethasone, prednisolone and mifepristone, the antiinflammatory and metabolic effects are similarly weak or strong. In contrast, with RU 24858, they are clearly distinct, suggesting that this drug can selectively trigger the antiinflammatory glucocorticoid effect, without triggering the side effects on metabolic regulation.
The metabolic effects of glucocorticoids include increased protein breakdown and excessive glucose formation, leading to loss of muscle mass, osteoporosis, and symptomatic diabetes, all of which are prominent side effects of prolonged systemic corticosteroid treatment. Therefore, the discovery of agonists that separate the two action mechanisms of glucocorticoids is really quite exciting!
More detailed studies on RU 24858 found that some of its antiinflammatory effects are, in fact, mediated by transactivation, not transrepression [62,63]. Like the thyroid hormone receptors, the glucocorticoid receptor can form homodimers as well as heterodimers with other nuclear hormone receptors; it seems possible that RU 24858 promotes formation of some, but not all of these receptor dimers.
While the term “dissociated steroids” has been used to describe the activity of compounds like RU 24858, we already know a better name for it—this is really just another case of agonist-specific coupling, and indeed a more compelling and practically relevant one than the examples we have seen with G protein-coupled receptors.
| 7.4.6 |
11-β-Hydroxysteroid dehydrogenase prevents non-specific mineralocorticoid receptor activation by cortisol |
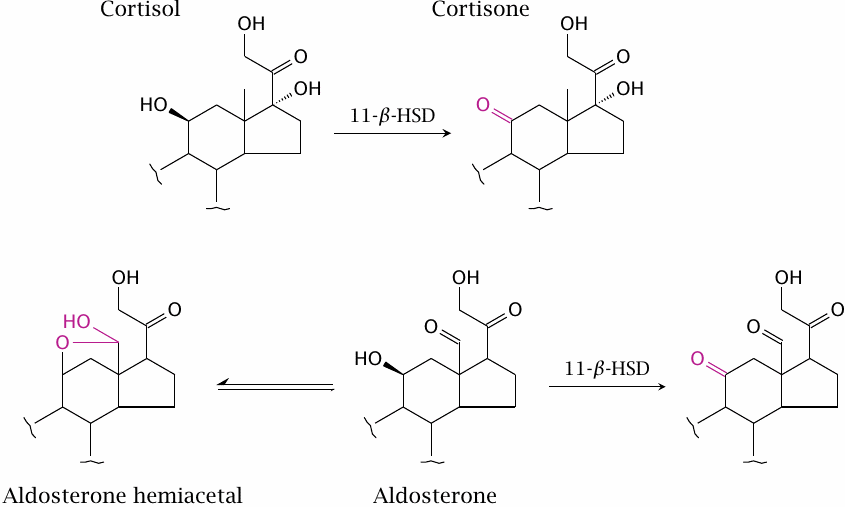
The mineralocorticoid receptor (MR) is expressed in the tubular epithelia of the kidney (see slide 3.7.4). When activated, the receptor induces the expression of transporters that mediate excretion of potassium and reuptake of sodium; it therefore plays an important role in the homeostasis of these two electrolytes that are of paramount importance in cell excitation. Mineralocorticoid activity may become excessively high downstream of other physiological dysregulation in chronic heart disease, and as a side effect of excess glucocorticoid activity, since glucocorticoids may also have some activity on the MR.
When studied in vitro, cortisol and aldosterone activate the MR to a similar degree. However, in vivo, activation of the MR by cortisol is inhibited by the enzyme 11-β-hydroxysteroid dehydrogenase (11-β-HSD), which is strongly expressed in kidney cells along with the MR. The enzyme dehydrogenates cortisol to cortisone, which is inactive at the MR. Aldosterone is protected from dehydrogenation by 11-β-HSD because it mostly exists in the hemiacetal form.
| 7.4.7 |
Mineralocorticoid receptor ligands |
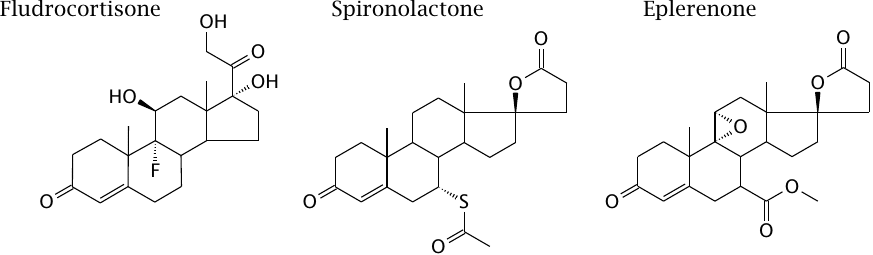
Fludrocortisone is a mineralocorticoid receptor agonist; it may be used in substitution treatment in patients who lack normal aldosterone synthesis. The antagonists spironolactone and eplerenone are used in cardiac insufficiency and other states of excessive endogenous mineralocorticoid activity.
| 7.4.8 |
Gonadal biosynthesis of androgens and estrogens |
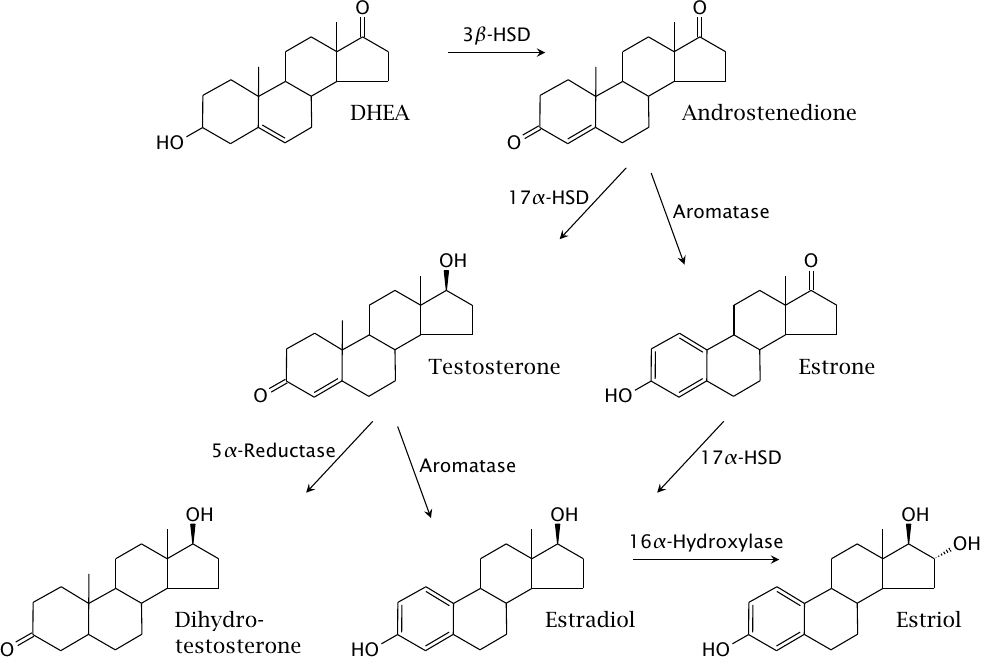
Abbreviations: DHEA, dehydroepiandrosterone; 3β-HSD and 17α-HSD, 3β- and 17α-hydroxysteroid dehydrogenase, respectively. Aromatase and 16α-hydroxylase are cytochrome P450 enzymes. Estradiol, estriol and estrone are estrogens; all others are androgens.
Gonadal synthesis of androgens and estrogens starts with dehydroepiandrosterone, which is supplied by the adrenal glands but can also be locally formed in the testicles and ovaries. Note that all estrogens are derived from androgens, and that aromatase is required for their synthesis. Accordingly, aromatase inhibitors are used in the treatment of estrogen-dependent gynecological tumors (see slide 12.4.2).
Dihydrotestosterone is the most potent androgen; its specific activity is five times higher than that of testosterone. Therefore, inhibition of 5α-reductase, which converts testosterone to dihydrotestosterone, significantly reduces the level of androgen activity. This is exploited in the treatment of androgen-dependent prostate tumors.
| 7.4.9 |
Synthetic analogues of gonadal steroids |
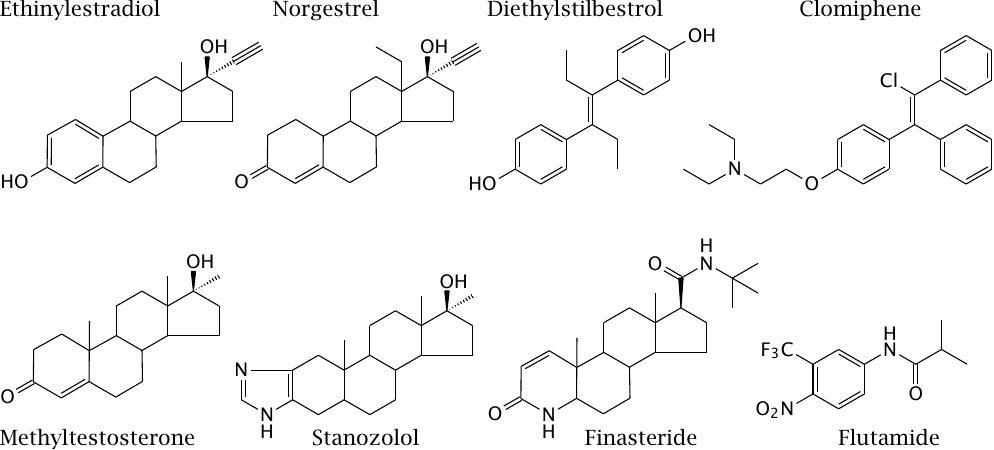
Natural gonadal steroids are subject to strong first-pass effects in the liver and therefore cannot be used orally. Ethinylestradiol, norgestrel and methyltestosterone are orally available estrogen, progestin and androgen analogues, respectively. Diethylstilbestrol was an early synthetic estrogen but was withdrawn after it was found to induce cancer.
The main use of synthetic estrogens and progestins is for contraception in females. As with glucocorticoids or thyroid hormones, the production of endogenous estrogens and progestins is controlled by the hypothalamic-pituitary axis, and these steroids exercise feedback inhibition on the hypophyseal secretion of follicle-stimulating hormone (FSH) and luteinizing hormone (LH). Introducing extrinsic estrogens and progestins suppresses the secretion of FSH and LH, which deprives the ovaries of their hormonal stimulus and thus prevents ovulation.
In men, FSH and LH support sperm cell formation (spermatogenesis). Most efforts devoted to male contraception follow the same general idea as that used in females, namely, the suppression of FSH and LH secretion through application of synthetic androgens. While this works in principle, most clinical studies so far have concluded that behavioral or medical side effects were not acceptable.
Clomiphene is a “selective estrogen receptor modulator;” this class of compounds acts variously agonistic and antagonistic in different tissues, which must ultimately again arise from some sort of agonist-specific coupling. It inhibits the negative feedback exercised by gonadal steroids on the hypothalamus and the hypophyseal gland, and therefore increases the release of gonadotropin-releasing hormone (GnRH) from the hypothalamus and subsequently LH and FSH from the hypophyseal gland. It is used in fertility treatment in women, sometimes resulting in multiple pregnancies. Stanozolol is an oral androgen that is popular among body builders.60
Finasteride is a 5α-reductase inhibitor; it prevents conversion of testosterone to the considerably more active dihydrotestosterone and therefore reduces androgen activity. Flutamide is an androgen receptor antagonist. Both drugs are used in the treatment of hormone-dependent prostate cancer.
| 7.5 |
Endocrine control of bone mineralization |
Bone matrix consists of structural proteins, mostly collagen, and bone mineral, which is mostly hydroxyapatite (Ca5(PO4)3OH). In addition to its structural role, bone mineral also serves as a reservoir for calcium and phosphate. Its deposition and mobilization are controlled by several hormones.
The most widespread affliction of bone matrix is osteoporosis. In this condition, which is most common among elderly women, both the matrix proteins and the bone mineral are depleted. The most effective treatment is substitution of estrogens, the endogenous levels of which recede after menopause; estrogens are required for sustaining bone mass.61 However, estrogen substitution is fraught with a considerable risk of tumor promotion that increases with the cumulative duration of therapy. Other forms of treatment address the bone mineral balance, with limited success.
| 7.5.1 |
Formation and resorption of bone matrix |
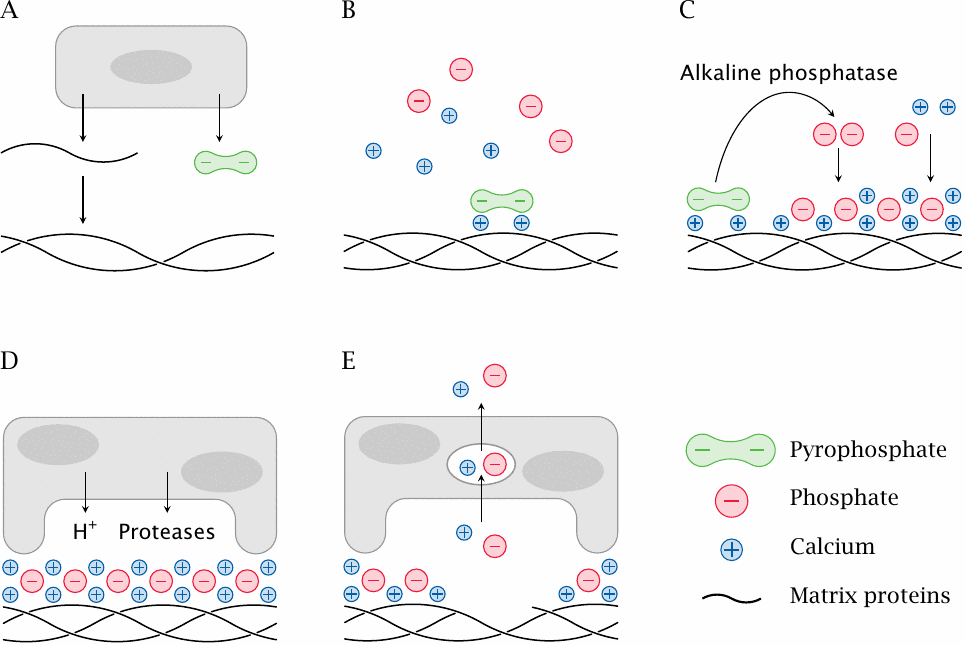
While bone matrix is evidently very durable after death, it is quite dynamic in life. Bone matrix is formed by one specialized cell type, the osteoblasts, and dissolved by another one, the osteoclasts.
- (A)Osteoblast cells secrete bone matrix proteins that self-assemble into fibrils. They also produce and release pyrophosphate ions.
- (B)The bone matrix proteins provide nucleation sites for the deposition of bone mineral crystals. Pyrophosphate binds to incipient bone mineral crystals and inhibits their premature growth.
- (C)Alkaline phosphatase, which is also produced by osteoblasts, cleaves the pyrophosphate. This triggers the deposition of bone mineral (hydroxyapatite).
- (D)Osteoclasts dissolve bone matrix. They attach to the surface of the bone matrix and seal off a patch of surface underneath. Into the occluded pocket, they secrete acid that dissolves the bone mineral, as well as proteases that digest the embedded matrix proteins.
- (E)Calcium and phosphate ions are taken up by the osteoclasts via endocytosis and then released into the circulation.
Osteoblasts and osteoclasts are both continuously active. Their relative levels of activity are under hormonal control and determine whether there will be a net accumulation or dissolution of bone matrix and minerals.
| 7.5.2 |
Hormones that affect bone mineralization and bone matrix |
-
Parathyroid hormone (PTH)
- mobilizes calcium and phosphate from the bone
- promotes calcium retention and phosphate elimination in the kidneys
- promotes activation of vitamin D by hydroxylation
- Calcitonin: promotes deposition of calcium and phosphate in the bone
-
Calcitriol (activated vitamin D)
- promotes intestinal calcium and phosphate uptake
- inhibits PTH secretion
- Estrogens: sustain bone matrix
Parathyroid hormone (PTH) is a peptide hormone that is secreted by the parathyroid glands, which are four little cell clusters attached to the thyroid gland. Calcitonin is secreted by the C cells in the thyroid gland itself (see slide 7.3.2). Both hormones are regulated by the blood calcium level; low blood calcium stimulates release of PTH and inhibits calcitonin secretion, and high calcium does the opposite.
While PTH and calcitonin manage the short term calcium homeostasis, calcitriol is important in long term control. Estrogens affect not only the bone mineral but also the bone matrix density.
| 7.5.3 |
Endogenous biosynthesis of calcitriol |
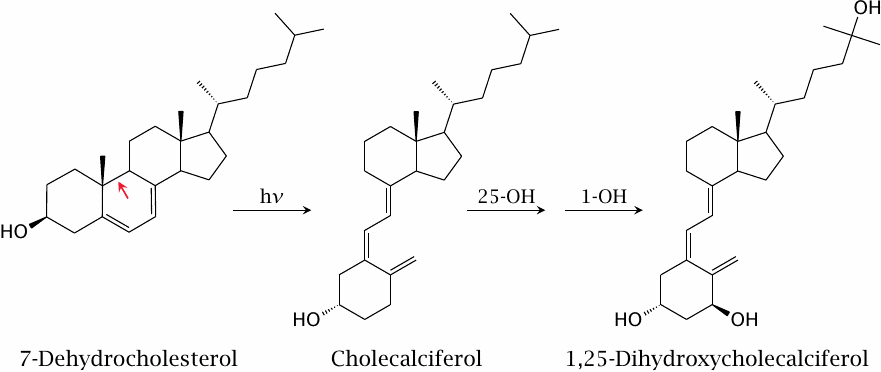
While conventionally referred to as a vitamin and often used as a supplement, calcitriol can actually be synthesized from scratch in human metabolism. The pathway branches off from cholesterol biosynthesis at 7-dehydrocholesterol, which is also the immediate precursor of cholesterol. The initial reaction is not enzyme-catalyzed but requires a UV photon.
The slide summarizes the synthetic pathway. Photochemical breakage of one bond in the ring (red arrow) in 7-dehydrocholesterol produces cholecalciferol. Two subsequent enzymatic hydroxylations yield 1,25-dihydroxycholecalciferol (calcitriol).62
| 7.5.4 |
Drugs that influence calcium balance and bone mineralization |
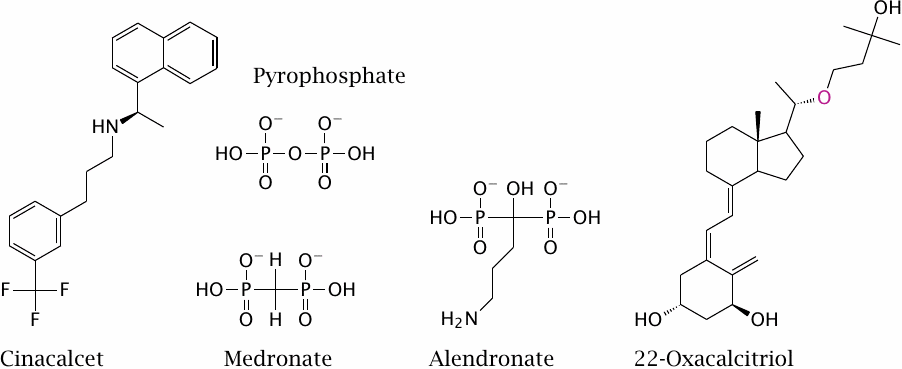
The secretion of PTH by the parathyroid gland is regulated by an unusual G protein-coupled receptor that senses the extracellular calcium level. Cinacalcet is an allosteric agonist of this receptor; its effect is to downregulate PTH secretion and thereby preserve bone minerals.
The bisphosphonates medronate and alendronate inhibit the resorption of bone matrix by osteoclasts. Like pyrophosphate—which is not a drug, but shown just for comparison—they bind to the surface of bone mineral particles. When mobilized by acid secreted from an osteoclast, they undergo endocytosis; in this way, they are preferentially targeted to the osteoclast cells. Within the cells, they inhibit the synthesis of farnesylpyrophosphate (see next slide).
22-Oxacalcitriol is an analog of calcitriol. Like calcitriol, the drug inhibits parathyroid hormone secretion and thereby reduces calcium mobilization from the bone matrix. Unlike calcitriol, however, 22-oxacalcitriol does not greatly increase intestinal calcium uptake.63
| 7.5.5 |
Sites of action of statins and bisphosphonates |
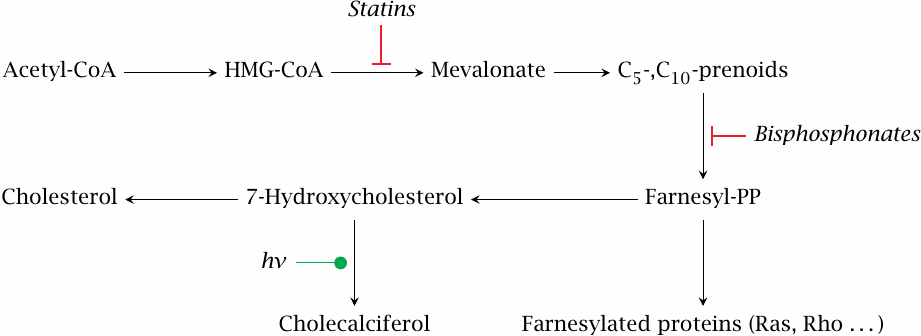
Bisphosphonates inhibit the formation of farnesylpyrophosphate, which is not only an intermediate in sterol synthesis but also a cosubstrate in the posttranslational modification of some membrane-associated proteins. Inhibited protein farnesylation causes apoptosis of the osteoclast cells.64
In the sterol synthesis pathway, both statins and bisphosphonates act upstream not only of protein farnesylation but also of cholecalciferol synthesis. While the bisphosphonates affect mainly osteoclasts, the statins indeed inhibit cholesterol synthesis in the liver, and one would therefore expect them to lower vitamin D levels. However, with patients on atorvastatin, the opposite has been reported [66]; and moreover, clinical studies support a beneficial effect of statins in osteoporosis [67]. This example clearly shows that, as the saying goes, the difference between theory and practice is greater in practice than in theory.