| 5 |
G protein-coupled receptors |
| 5.1 |
Overview |
As already stated earlier (slide 1.2.3), G protein-coupled receptors (GPCRs) form the largest class of drug targets in the human body. It is therefore appropriate to examine and understand them in some detail.
The human genome contains genes for several hundred GPCRs. For a sizable number of these receptors, the ligands and physiological functions are not yet known. Once these “orphan” receptors will have become “deorphanized”,28 at least some of them will probably become drug targets also.
| 5.1.1 |
Drugs that act on G protein-coupled receptors: Some examples |
| Drug | Major receptor | Drug action | Clinical use |
| salbutamol | β2-adrenergic | partial agonist | bronchodilation |
| fexofenadine | histamine H1 | inhibitor | antiallergic |
| atropine | muscarinic | inhibitor | pupil dilation |
| haloperidol | dopamine | inhibitor | antipsychotic |
| morphine | opioid | agonist | pain killer |
| losartan | angiotensin | inhibitor | antihypertensive |
| clopidogrel | adenosine | inhibitor | anticoagulation |
These selected examples suffice to illustrate the wide-ranging and diverse roles of GPCRs in physiology and pharmacology.
| 5.1.2 |
GPCR structure |
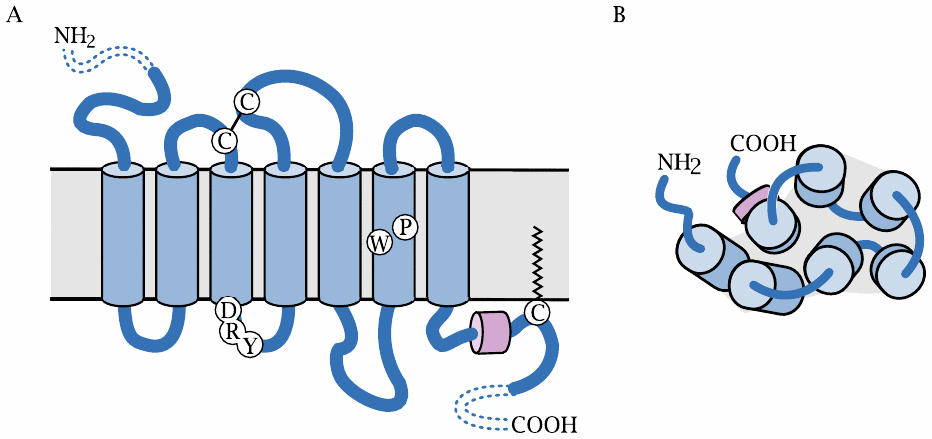
All GPCRs (with the exception of rhodopsin) are located in the cytoplasmic membrane. The snake diagram on the left shows the arrangement of the seven transmembrane helices: the N-terminus faces outward, and the C-terminus is located inside the cell. The diagram also indicates the location of several amino acid residues that are conserved within the largest subfamily of GPCRs, and which play key roles in receptor activation. These include the DRY motif at the cytoplasmic end of helix 3, the proline and tryptophan residues in helix 6, and the short, horizontally positioned C-terminal helix 8.
The schematic on the right shows the view from the extracellular side. The helices are arranged in an approximately circular fashion within the membrane plane. With many receptors, the crevice in the middle of the helices contains the ligand binding site, which is accessible from the extracellular side.
While the extracellular N-terminal domain is short in most GPCR molecules, some receptors have larger N-terminal domains that may contain ligand binding sites or other functional features. We will see some examples later.
| 5.1.3 |
The G protein cycle |
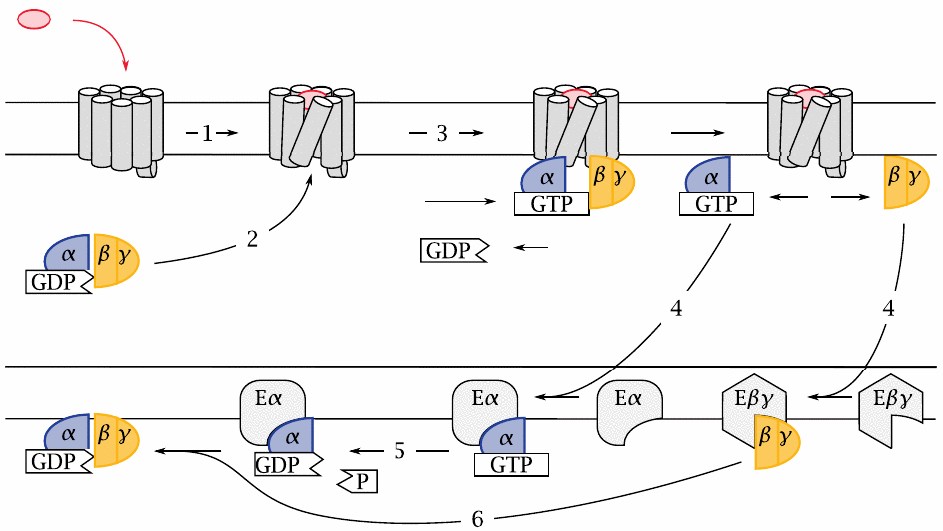
GPCRs activate GTP-binding proteins, or G proteins for short, which in turn activate various effector proteins. G proteins are heterotrimers; their subunits are referred to by the Greek letters α, β and γ.
G proteins undergo repeated cycles of activation and inactivation. The cycle starts when an agonist binds to the extracellular face of a GPCR (1), which changes the conformation of the entire GPCR molecule, including its intracellular surface. In this activated conformation, the GPCR binds a G protein (2). The α-subunit of this G protein then releases a molecule of GDP, which was left behind by a previous turn of the cycle. Next, it binds GTP and then dissociates from the βγ-dimer. The two dissociated G protein fragments are now free to seek out and bind to their respective effector proteins (4), which have various biochemical activities (see later).
After some time has elapsed, the bound GTP molecule is cleaved by the built-in GTP′ase activity of the Gα-subunit (5). This causes the α-subunit to leave its effector and to again associate with a Gβγ dimer (6). The inactive trimer then awaits another round of activation.
| 5.2 |
G protein binding and dissociation can be observed with GFP fusions |
The experiments described in the following illustrate how the activities of GPCRs and of G proteins can be observed within cells using the green-fluorescent protein from Aequorea jellyfish and its recombinant derivatives. These methods are straightforward and commonly used today; they were, however, not yet available in the early studies that first discovered how these proteins work.
| 5.2.1 |
The fluorophore in green-fluorescent protein forms autocatalytically |
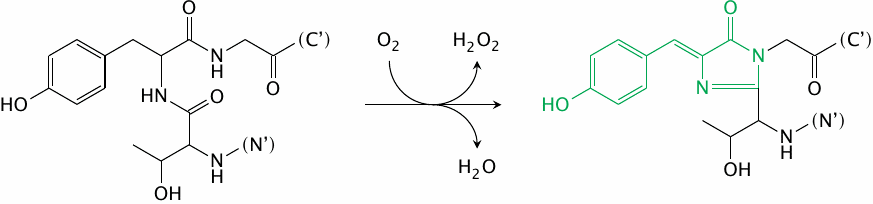
The great power of the green-fluorescent protein (GFP) is due to its entirely spontaneous formation of a fluorophore—it needs no help from any other protein, and it therefore can easily be used in almost any type of cell or cellular compartment. GFP is typically used through translational fusion with a protein of interest.
While the wild-type GFP is excited with ultraviolet light, several mutants have been created that can be excited at different wavelengths within the visible spectrum, and which can be combined with one another for fluorescence resonance energy transfer (FRET) experiments. In the mutant cyan-fluorescent protein (CFP), the tyrosine residue that becomes part of the fluorophore is replaced by tryptophan. The yellow-fluorescent protein (YFP) retains the unmodified fluorophore of GFP; it contains a mutant tyrosine residue that is not part of the fluorophore, but engages in π-stacking interactions with the latter and thereby shifts its absorption and emission spectra.
| 5.2.2 |
FRET detection of G protein binding to adenosine receptors |
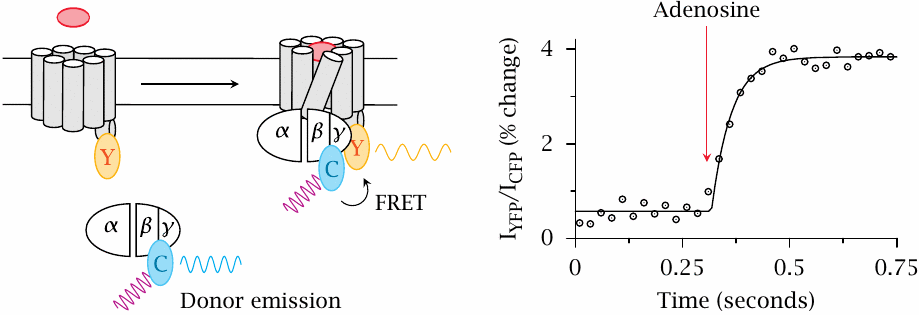
In this experiment, the yellow-fluorescent protein (Y) was translationally fused to the A2A adenosine receptor, and the cyan-fluorescent protein (C) to the γ-subunit of the cognate G protein (Gs). Addition of adenosine activates the receptor and triggers binding of Gs to the receptor. The ensuing FRET from CFP to YFP is detected through an increase in the ratio of YFP emission to CFP emission.
The observed change in the extent of FRET is fairly small, suggesting that only a small fraction of the available G protein molecules associates with the activated receptor. Figure prepared from original data in [30].
| 5.2.3 |
FRET detection of G protein dissociation |
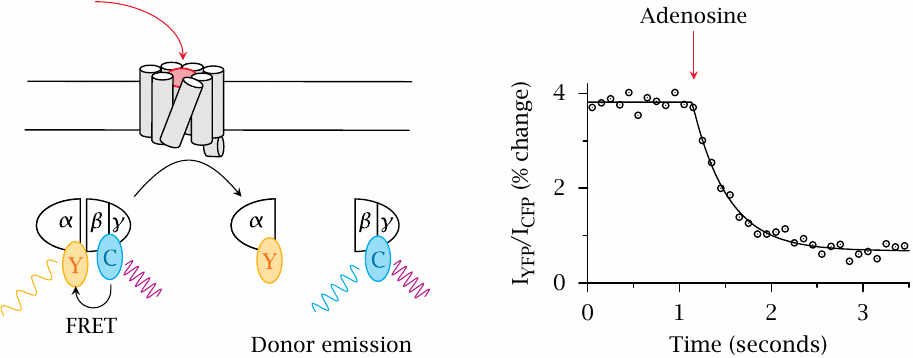
In this experiment, FRET between CFP and YFP was used to detect the dissociation of the heterotrimeric G protein. In this case, the extent of FRET decreases upon receptor activation. Again, the change in FRET is small.
Note the difference in time scales between this slide and the preceding one: Dissociation of the G protein is noticeably slower than its binding to the activated receptor. Figure prepared from original data in [30].
| 5.3 |
G protein effector mechanisms |
For each of the three subunits of the heterotrimeric G proteins—α, β and γ—there are several subtypes, which may combine into heterotrimers in various permutations. However, overall, G proteins are less diverse than GPCRs; therefore, multiple types of GPCRs must converge upon the same G proteins and trigger the same intracellular responses.29 We will now look at the major intracellular signaling cascades triggered by different G proteins.
| 5.3.1 |
Adenylate cyclase |
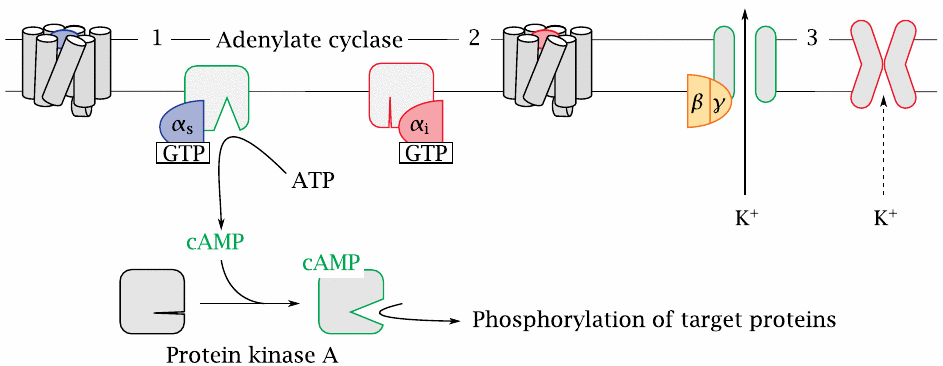
One important effector protein is adenylate cyclase. This membrane-associated enzyme converts ATP to cyclic AMP (cAMP), which is an allosteric activator of protein kinase A.
Adenylate cyclase is controlled by two different Gα subunits, which are activated by different GPCRs. The stimulatory α-subunit (Gαs, 1) activates the enzyme, whereas the inhibitory α-subunit (Gαi, 2) inhibits it.
An important effector molecule for Gβγ dimers are K+ channels of the Kir type (3). Opening of these channels will cause hyperpolarization of the cell membrane. In excitable cells, this will tend to reduce the level of activity.
| 5.3.2 |
The phospholipase C cascade |
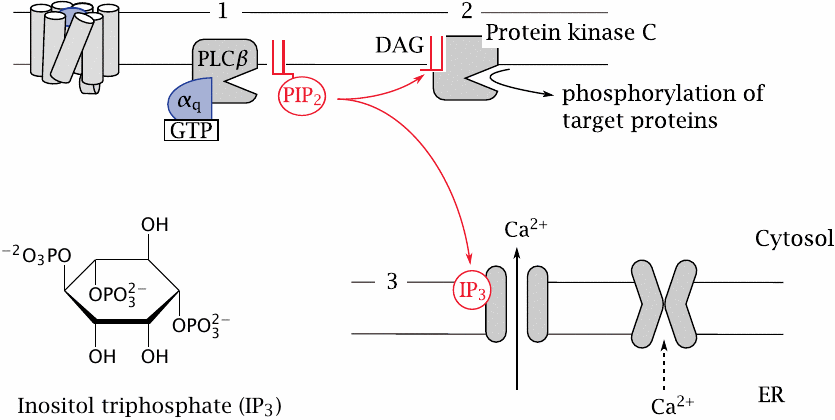
Another effector pathway that is activated downstream of many pharmacologically important GPCRs is the phospholipase C cascade. For example, the contraction of vascular smooth muscle cells downstream of α-adrenergic receptors (slide 2.3.4) or angiotensin receptors (slide 1.2.5) is mediated by this pathway.
In this cascade, the α-subunit of Gq stimulates phospholipase Cβ (1). The activated enzyme then cleaves a specific membrane lipid, phosphatidylinositol-bisphosphate (PIP2), into two secondary messengers, namely, diacylglycerol and inositoltriphosphate.
Diacylglycerol (DAG) is a very hydrophobic molecule that remains associated with the cytoplasmic membrane, where it activates protein kinase C (2). Phosphorylation of target proteins by this kinase causes various downstream effects.
The other messenger, inositoltriphosphate (IP3), is water-soluble. It travels through the cytosol to the ER membrane, where it activates the IP3 receptor, which is a calcium channel (3). Ca++ ions released from the ER into the cytosol activate calmodulin, which in turn will affect numerous calmodulin-dependent proteins. In smooth muscle, a calmodulin-dependent protein kinase phosphorylates myosin, which triggers contraction.
| 5.3.3 |
Summary of G protein effector mechanisms |
| Class | Effectors and Effects | Some activating GPCRs | |
| Gαs | stimulation of adenylate cyclase (various types) | β-adrenergic, 5-HT4, 5-HT6, 5-HT7, D1, D5; ACTH | |
| Gαi/o | inhibition of adenylate cyclase; activation of extracellular signal-regulated kinase (ERK) | α2-adrenergic, 5-HT1, D2, D3, D4 | |
| Gαq/11 | stimulation of Phospholipase Cβ (various subtypes) | α-adrenergic, 5-HT2, H1, GABAB | |
| Gα12/13 | indirect activation of RhoA GTPase and of phospholipase A2 | 5-HT4, AT1, protease- activated receptors |
The activities of the first three G proteins listed here were illustrated in the preceding two slides. The enumeration of activating receptors for each of these cascades is neither exhaustive nor recommended for memorization, but only intended to illustrate the convergence of different receptors on the same G proteins.
One effect that occurs downstream of the fourth listed G protein, Gα12/13, is the activation of phospholipase A2, which in turn promotes the formation of eicosanoids (see chapter 9). The protein also activates RhoA GTPase, which influences transcriptional regulation.
A single GPCR may interact with multiple G proteins, which may set off different downstream cascades. Occasionally, different receptor agonists may activate various downstream cascades to different extents, which is referred to as agonist-specific coupling (see slide 2.5.4).
| 5.4 |
Structure and function of GPCRs |
Much experimental effort has been devoted to studying the conformational changes that are at the heart of GPCR function. Many of these studies have been performed on rhodopsin. This molecule differs from other GPCRs in being activated not by ligand binding and dissociation, but instead by photoisomerization of its covalently bound retinal chromophore. However, it is the easiest GPCR molecule to obtain in abundance, and it was the first one to be crystallized, which made it an attractive model. Since it shares extensive homology with many GPCRs that are drug targets, it is also a credible and useful model.
Crystal structures of other GPCRs have now been experimentally determined, and they will indubitably enhance our understanding and the precision of drug design. However, crystal structures are static, and other experimental methods remain relevant for understanding the molecular movements and interactions. We will consider a few selected examples below. The first example involves the receptor for substance P, a peptide neurotransmitter.
| 5.4.1 |
Substance P and its competitive antagonist CP-96345 |
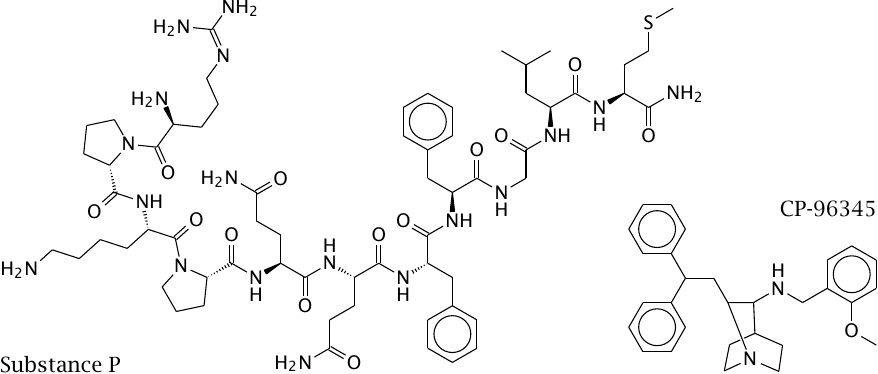
Substance P, also known as neurokinin 1 (NK1), is a peptide neurotransmitter that is involved in the perception of pain and in triggering emesis. NK1 receptor antagonists such as CP-96345 are being developed for controlling chemotherapy-induced emesis in cancer patients.
Substance P and CP-96345, as well as eledoisin, another peptide that is related to substance P, were used in the following experiment that was designed to determine the ligand binding sites of the NK1 and NK3 receptors.
| 5.4.2 |
Using receptor chimeras to locate the ligand binding sites of NK receptors |
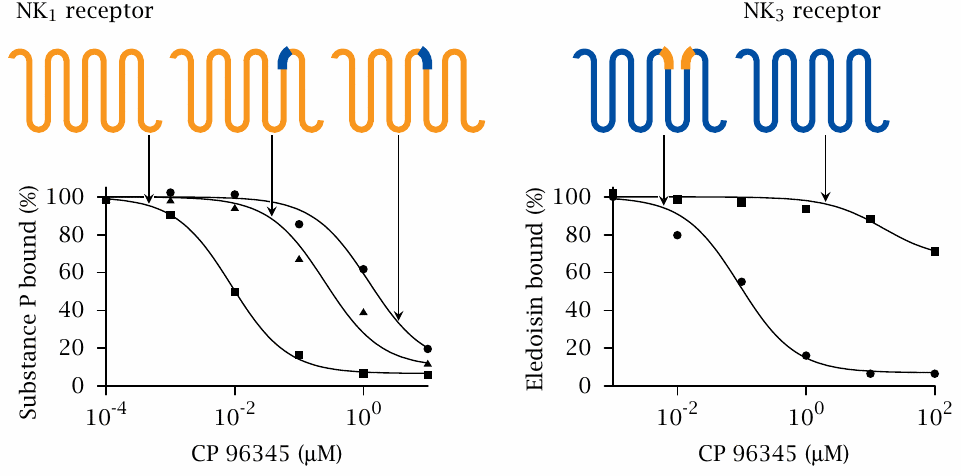
CP-96345 displaces substance P from the wild-type NK1 receptor (left) but does not effectively compete with eledoisin at the NK3 receptor (right). When either of two short stretches located at the extracellular ends of transmembrane helices 5 or 6, respectively, in NK1 is replaced by the homologous sequences from NK3, the inhibitory potency of CP-96345 is greatly reduced. This indicates that binding of CP-96345 to the chimeric receptors is disrupted.
The reciprocal exchange markedly increases the sensitivity of the NK3 receptor to inhibition by CP-96345. Therefore, the short sequence stretches that were exchanged between the receptors carry determinants that are crucial for binding CP-96345. Note that the chimeric receptors retain affinity for substance P and eledoisin, respectively. Figure prepared from original data in [31].
| 5.4.3 |
Engineered disulfide bonds pinpoint helix movements involved in GPCR function |
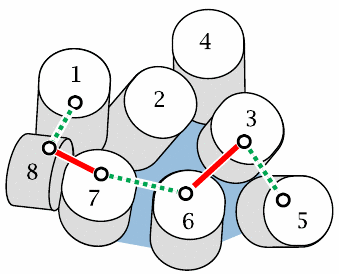
This slide is supposed to show a view onto the cytoplasmic surface of rhodopsin. Lines between helices indicate disulfide bonds that were engineered into the molecule one by one. This is done by substituting pairs of amino acid residues in strategic locations with pairs of cysteines. If two cysteines are sufficiently close in space, they will form a disulfide bond spontaneously.
Bonds drawn with solid red lines prevent receptor activation, whereas those shown as dashed green lines do not. These findings indicate that helices 7 and 8, as well as helices 3 and 6, must be able to move relative to one another in order to allow receptor activation. Figure based on reference [32].
| 5.4.4 |
Protonation of residue E134 of rhodopsin in response to light stimulation |
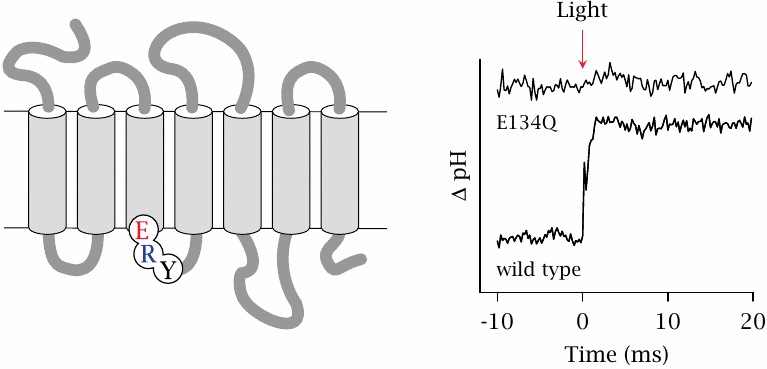
The cytosolic end of helix 3 of rhodopsin contains a functionally crucial tripeptide sequence, consisting of glutamate in position 134, followed by arginine and tyrosine. This sequence motif—with aspartate often taking the place of glutamate, see slide 5.1.2—is conserved in the entire group of rhodopsin-like receptors, which are the largest subclass of GPCRs. From the disulfide tethering experiment just discussed, we already know that this location is important in receptor activation. The experiment shown here looks at the role of the conserved ERY motif.
Purified and detergent-solubilized rhodopsin was suspended in unbuffered water, so as to maximize the pH change in response to a small change in the number of free protons, such as can be expected to occur through protein protonation or deprotonation. When wild type rhodopsin is exposed to a flash of light, the pH value jumps up, indicating uptake of protons by the protein. This protonation occurs at residue glutamate 134, as is shown by its absence in a mutant (E134Q) that contains glutamine instead of glutamate in this position.
A plausible explanation is that protonation of E134 breaks a salt bridge with the adjacent arginine and thereby removes an energetic constraint that ties rhodopsin down in its inactive conformation. Figure prepared from original data in [33].
| 5.5 |
Protease-activated GPCRs |
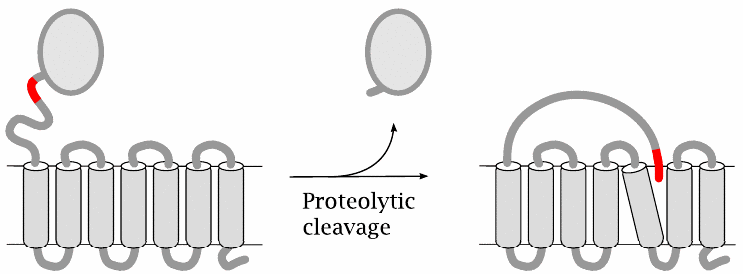
Protease-activated receptors contain an inhibitory domain that extends the extracellularly exposed N-terminus. A receptor is activated when a protease cleaves this domain and thereby exposes a built-in receptor ligand, which then associates non-covalently with a ligand binding site located in the transmembrane domain of the same receptor molecule.
The location of this binding site, and the events set in motion by the binding of the intramolecular ligand, are similar to those found in other, conventional GPCRs. However, there is one important difference: While activation of other GPCRs is reversed when the ligand is diluted, activation of protease-activated receptors is irreversible, since the ligand is part of the receptor and not amenable to dilution. Protease-activated receptors therefore can only be inactivated by endocytosis and degradation (see slide 5.7.1).
| 5.5.1 |
Pharmacological inhibition of protease-activated receptors |
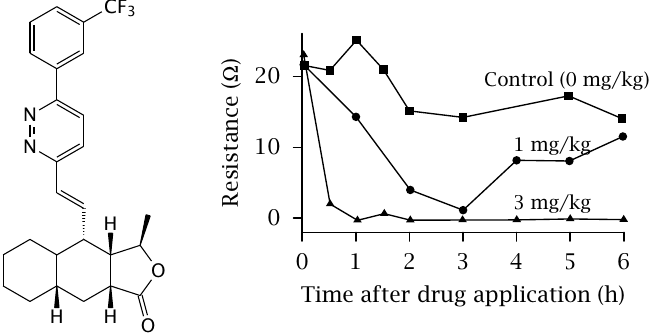
The protease-activated receptor PAR1 is found on thrombocytes and is activated by thrombin; it contributes to the activation of thrombocytes (see slide 10.6.7). Generally speaking, inhibition of thrombocyte activity is an important strategy in the prevention of myocardial infarction and stroke in patients with advanced atherosclerosis. Protease-activated receptors are interesting as a potential target in this application also.
The molecule shown on the left is a synthetic PAR1 inhibitor. Its activity as an inhibitor was assessed in the animal experiment summarized in the plot. The inhibitor was applied at time 0, and plasma samples were obtained at various times after application. The samples were then spiked with a peptide agonist of PAR1 in order to activate the receptor.30
Thrombocyte aggregation was measured through the electrical resistance between two electrodes immersed in the sample; as thrombocytes aggregate atop the electrode surfaces, the ohmic resistance between the electrodes increases. A decrease in resistance therefore indicates inhibited thrombocyte aggregation. Figure prepared from original data in [34].
| 5.6 |
GPCR oligomerization |
- Oligomers can comprise identical or different subunits
- Potential for cooperativity
- Potential for novel ligand specificity
- When receptors for antagonistic mediators form heterodimers, these mediators can “duke it out” already at the cell surface, reducing noise inside the cell
While GPCRs were initially believed to function as monomeric molecules, there is now solid evidence that many receptors form oligomers. While in some cases the functional effect of oligomer formation may amount to no more than a modicum of cooperativity in the response to the ligand, in others oligomerization has a more profound effect on receptor function; we will look at a few examples presently.31
| 5.6.1 |
Functional specialization in GABAB receptor heterodimers |
GABA (γ-aminobutyric acid) is a major inhibitory neurotransmitter in the brain. There are two major types of GABA receptors; the GABAA and GABAC receptors are ligand-gated channels, whereas the GABAB receptors belong to the GPCR family.
In contrast to most other GPCRs, GABAB receptors contain an extended N-terminal domain that is extracellularly located and also contains the ligand binding site. The receptors occur in various subtypes, which can form homo- and hetero-oligomers.
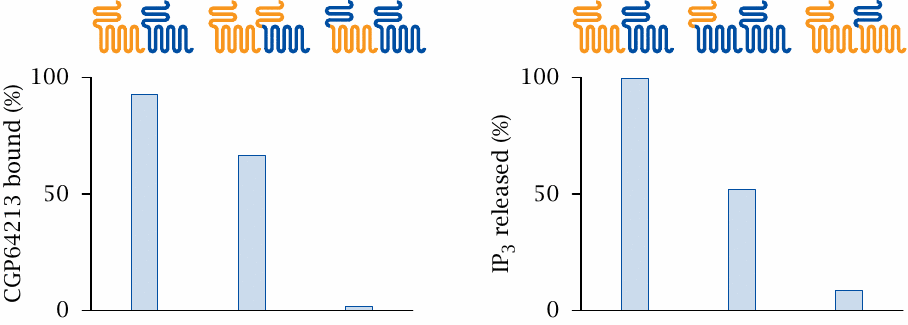
The study summarized here used chimeric receptors to examine the functional roles of the different subunits in GABAB-1/GABAB-2 receptor hetero-oligomers. Cells were transfected with both GABAB-1 receptor (orange) and GABAB-2 receptor (blue), or with combinations of either wild-type receptor with chimeric receptors as indicated.
In the experiment shown on the left, wild-type receptors were combined with chimeras so as to change all N-termini to either the GABAB-1 type (center) or the GABAB-2 type (right). When only GABAB-1 N-termini are present, ligand binding is somewhat diminished, but still considerable. In contrast, with only GABAB-2 N-termini present, ligand binding is abolished. Thus, in the heterodimer, the GABAB-1 subunit dominates ligand binding.
In the experiment on the right, all transmembrane domains were converted to either the GABAB-2 or the GABAB-1 type, and IP3 production downstream of receptor activation was measured. In this case, it is the GABAB-2 domain that on its own retains significant activity, while the GABAB-1 TM domain alone is inactive. Therefore, GABAB-2 dominates G protein activation. Figure prepared from original data in [35].
| 5.6.2 |
Bivalent agonists of muscarinic acetylcholine receptors |
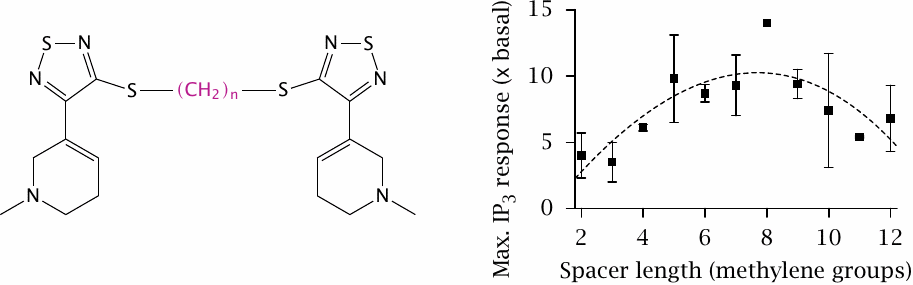
Like GABA, acetylcholine has receptors both among the ligand-gated channels and in the GPCR family. The latter are also referred to as muscarinic receptors (see Section 6.15). Muscarinic receptors form tetramers [36]. In the experiment depicted here, two molecules of the receptor ligand 1-methyl-1,2,5,6-tetrahydropyridyl-1,2,5-thiadiazole (phew!) were tied together with flexible spacers varying in length, and the extent of receptor activation was measured as IP3 release downstream of phospholipase C activation.
With increasing spacer length, receptor activation seems to go through an optimum and then to decrease again, suggesting that a certain amount of “tugging” at the tether may be conducive to activation.32 Figure prepared from original data in [37].
| 5.6.3 |
Novel receptor specificity: selective activation of κδ opioid receptor hetero-oligomers by a monovalent ligand |
Opioid receptors are activated by endorphins, which are peptide mediators, as well as by morphine and related natural and synthetic compounds. Opioid receptor agonists are very powerful pain killers, but as is well known, they are also inebriating and addictive. Separating these two effects is a desirable but elusive target.
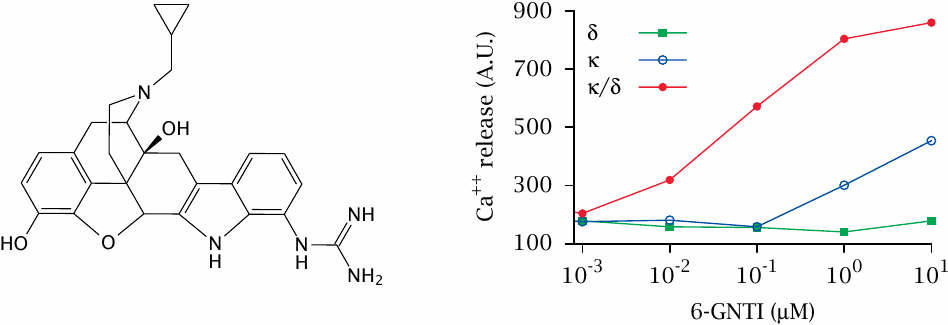
Opioid receptors have several subtypes that may form homo- or hetero-oligomers. Specific types of oligomers may be preferentially located in different areas of the central nervous system; for example, κδ-heterodimers primarily control pain conduction in the spinal cord.
Against this background, it may be more than a curiosity to observe that the drug 6-guanidinonaltrindole (6-GNTI) preferentially activates κδ-heterodimers. This was shown here through the determination of calcium release downstream of phospholipase C and IP3, using receptors recombinantly expressed in cell culture alone and in combination. Figure prepared from original data in [38].
| 5.6.4 |
Could receptor heterodimers be targeted with heterodimeric drugs? |
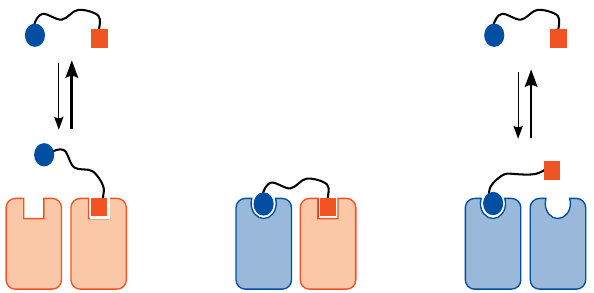
The discovery of the heterodimer-selective effect of 6-GNTI, while fascinating, was entirely fortuitous; it is currently impossible to rationally design monomeric drugs with such subtle specificity.
In order to rationally design heterodimer-specific drugs, one might consider the approach illustrated in this slide. While it would seem fairly obvious and straightforward, the devil is in the details. For this approach to work, the affinities of both half-ligands would have to be tuned such that they contribute about equally, and that divalent binding would be required for avid interaction with the receptor. An experimental study that attempts this approach is [39].
| 5.7 |
Feedback regulation of GPCR activity |
A GPCR will often be inactivated simply by dissociation of the agonist. However, some agonists may bind avidly and dissociate slowly, and with protease-activated receptors, activation is entirely irreversible. Therefore, additional mechanisms are needed to control receptor activity. One widely used mechanism for GPCR inactivation consists in phosphorylation.
| 5.7.1 |
GPCR deactivation by phosphorylation and endocytosis |
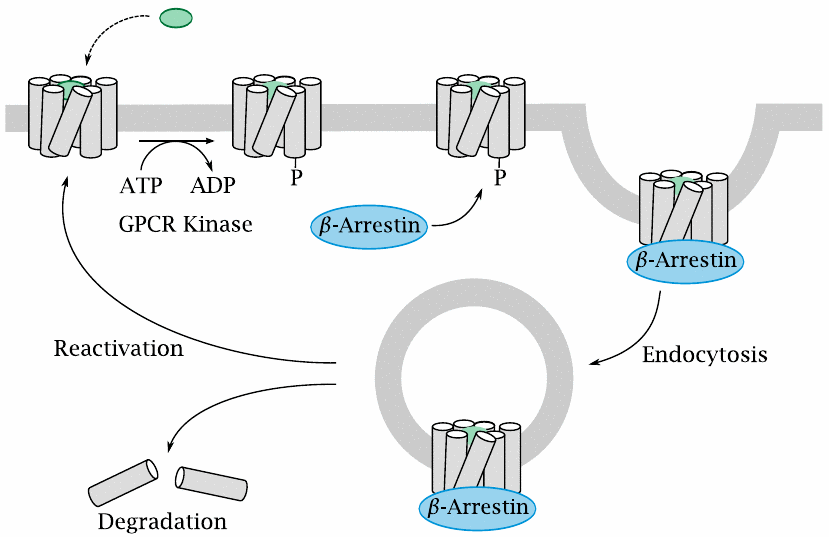
Activated GPCRs undergo phosphorylation by GPCR kinases. The phosphorylated GPCRs are then bound by β-arrestin, which prevents their further interaction with G proteins and also mediates their endocytosis. Endocytosed receptors may dissociate from β-arrestin and return to the cell surface, or alternatively they may undergo complete degradation within the endosome. As noted above, the latter fate always awaits protease-activated receptors.
GPCR phosphorylation and endocytosis are involved in tachyphylaxis, that is, a rapid partial desensitization to GPCR-activating drugs.
| 5.7.2 |
GPCR kinase 2 knockout attenuates tachyphylaxis of cardial β-receptors |
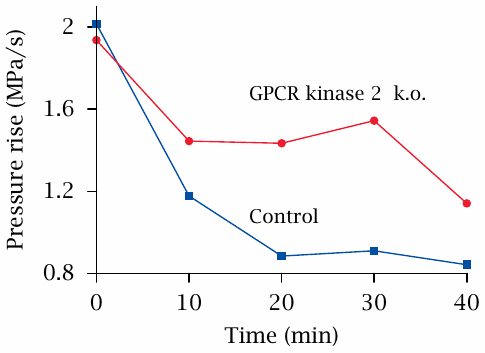
β-Adrenergic receptors are found in the heart, where they stimulate adenylate cyclase, which then increases the heart rate and strength of contraction; this, in turn, raises the blood pressure. In the control arm of the experiment shown here, application of the β-receptor agonist isoproterenol starting at time zero caused an increase in blood pressure, but the effect dropped by more than half within 20 minutes, despite continued application of the drug. This is an example of tachyphylaxis.
When GPCR kinase 2 is genetically knocked out, tachyphylaxis still occurs but is attenuated, indicating that this kinase participates in the inactivation of the β-receptors. Figure prepared from original data in [40].
| 5.7.3 |
Knock-down of arrestin may reduce GPCR-mediated signals |
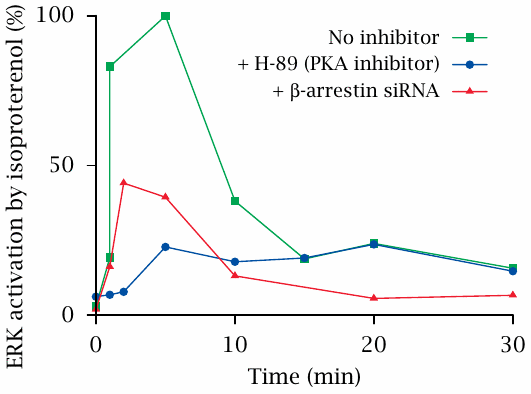
This experiment shows the activation of extracellular signal-regulated kinase (ERK) downstream of β-adrenergic receptors in response to isoproterenol. The experimental drug H-89 inhibits protein kinase A, which is activated downstream of β-receptors and adenylate cyclase.
ERK activation occurs in two phases. H-89 inhibits the early activation peak but not the subsequent, more shallow and protracted phase of ERK activation. This suggests that the late phase is mediated by a cAMP/PKA-independent mechanism.
Surprisingly, both the early and the late phase are inhibited when the expression of arrestin is suppressed with siRNA. This suggests that, in addition to its role in GPCR deactivation, β-arrestin may also transduce the signals represented by GPCR activation. There is indeed a growing body of evidence supporting this idea. Figure prepared from original data in [41].