| 9 |
Eicosanoids and related drugs |
| 9.1 |
Overview |
- “Local hormones”—sphere of action often limited to same anatomical site or tissue
- Involved in control of inflammation, fever, blood coagulation, pain perception, labor
- Derived from arachidonic acid and other polyunsaturated fatty acids, which occur in membrane phospholipids
- Most effects mediated by G protein-coupled receptors
- Some effects mediated by ion channels and nuclear hormone receptors
Eicosanoids are mediators that are derived from arachidonic acid several other polyunsaturated fatty acids. The precursor fatty acids are stored as parts of the phospholipid molecules that make up the membranes of the endoplasmic reticulum and the nucleus. From these phospholipids, the fatty acids are mobilized on demand by phospholipase A2 and then converted to the various mediators by a sequence of enzymatic modifications.
Among the many physiological functions that are regulated by eicosanoids, inflammation, pain perception and blood coagulation are of the greatest interest in pharmacotherapy.
| 9.1.1 |
Pathways and key enzymes in eicosanoid synthesis |
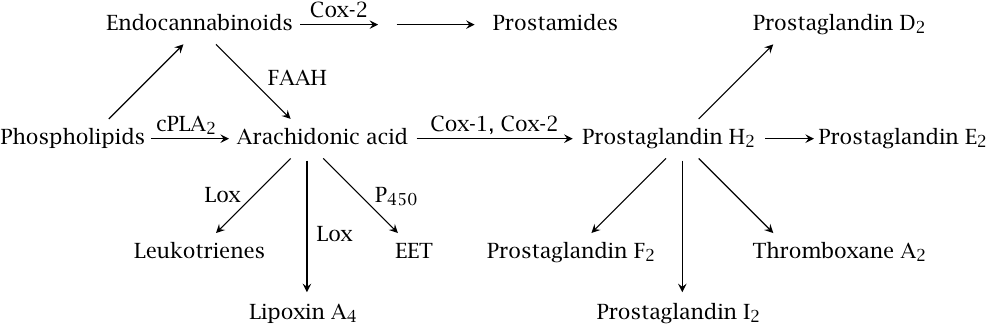
This slide introduces the major mediator classes and biosynthetic pathways. Release of arachidonic acid68 from membrane phospholipids by cytoplasmic phospholipase A2 (cPLA2) feeds cyclooxygenases 1 and 2 (Cox-1, Cox-2). These enzymes both form prostaglandin H2, which is then converted to other prostaglandins and to thromboxanes by specific synthases downstream.
Arachidonic acid is also the substrate of various lipoxygenases (Lox) and of some cytochrome P450 enzymes (P450), which produce leukotrienes, lipoxins, and epoxytrienoic acids (EET), respectively. Endocannabinoids, which are formed from arachidonate-containing phospholipids through separate pathways, are cleaved to arachidonic acid by fatty acid amide hydrolase (FAAH). Alternatively, they may be metabolized first by Cox-2 and subsequently by one of the prostaglandin synthases, giving rise to prostamides.
| 9.1.2 |
Calcium signals activate cPLA2 and initiate the synthesis of prostaglandins and leukotrienes |
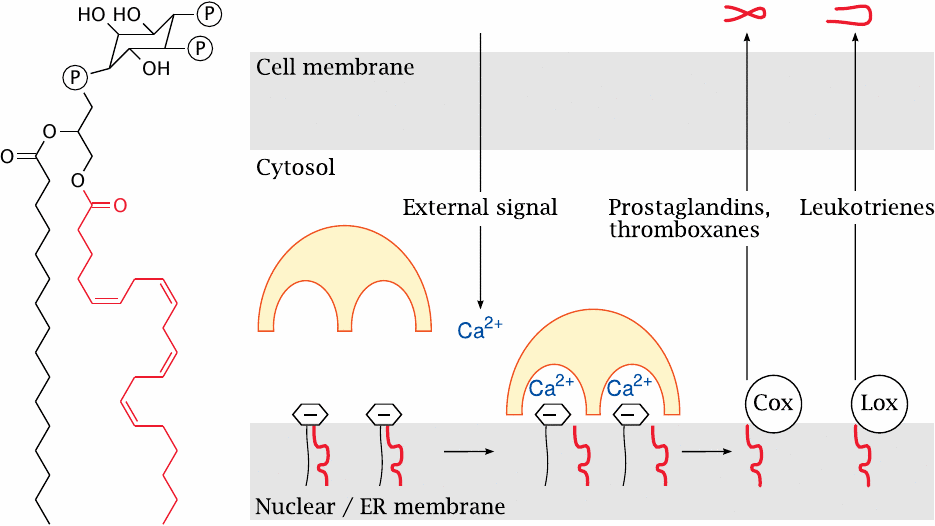
The major storage lipid for arachidonic acid is phosphatidylinositol-bis-phosphate (PIP2). In the structure of PIP2, the arachidonyl residue is highlighted.
Synthesis of prostaglandins and leukotrienes is triggered by increased levels of cytosolic calcium, which may for example be induced by the phospholipase C pathway downstream of GPCR activation. Calcium allows cytosolic PLA2 to bind and attack the negatively charged PIP2. The arachidonic acid thus released is converted to precursors of prostaglandins and thromboxanes by the membrane-associated cyclooxygenases (Cox), or alternatively to leukotriene precursors by lipoxygenases (Lox). These enzymes are found on the membranes of the nucleus and of the endoplasmic reticulum.
| 9.2 |
Biosynthetic pathways of prostaglandins and thromboxanes |
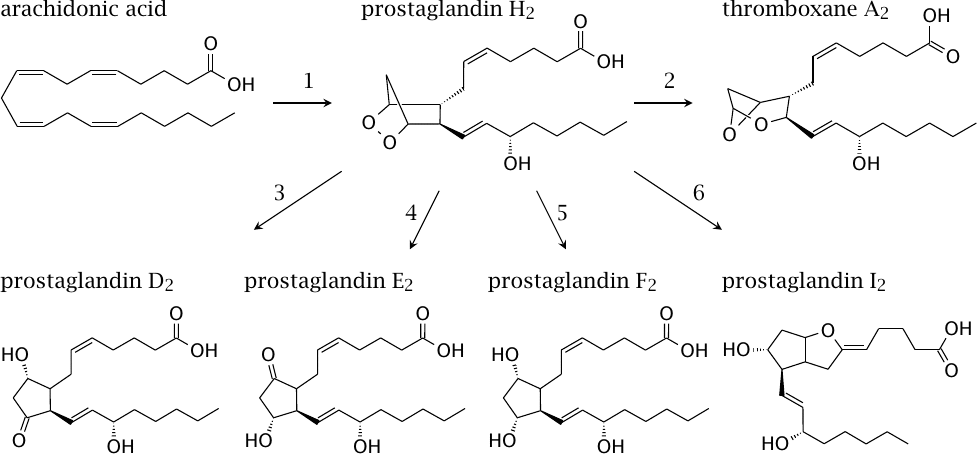
Among the mediators synthesized downstream of cyclooxygenase and prostaglandin H2, thromboxane A2 triggers thrombocyte aggregation, whereas prostaglandins E and I inhibit it (see slide 9.5.1). Prostaglandins D, E, F, and I are involved in pain perception, inflammation, and other physiological effects.
| 9.2.1 |
Eicosanoids synthesized by lipoxygenases |
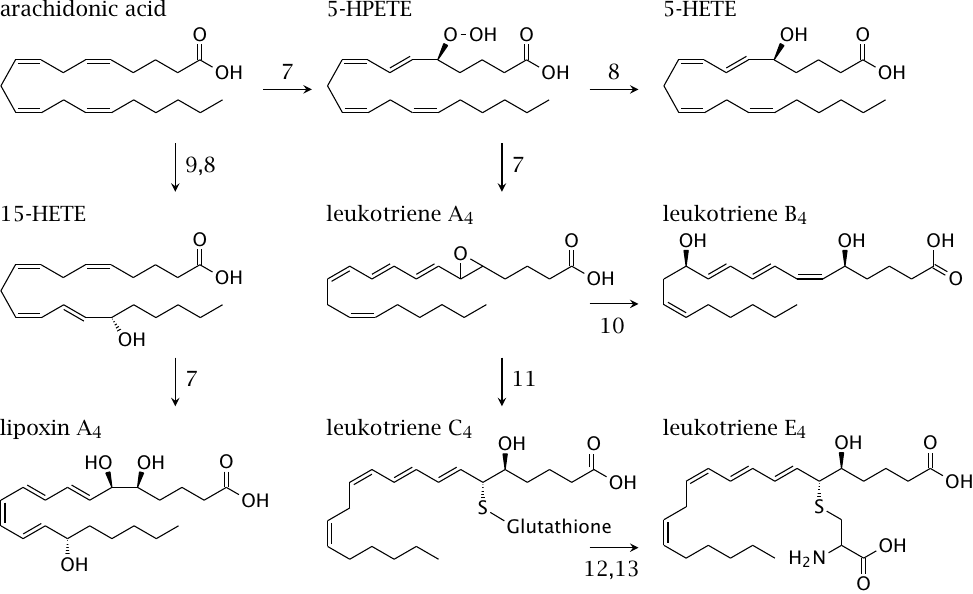
Leukotrienes are proinflammatory mediators; they play a major role in the pathogenesis of asthma. Lipoxins are important in the resolution of inflammation, that is, they are antiinflammatory.
| 9.3 |
The two steps of the cyclooxygenase reaction |
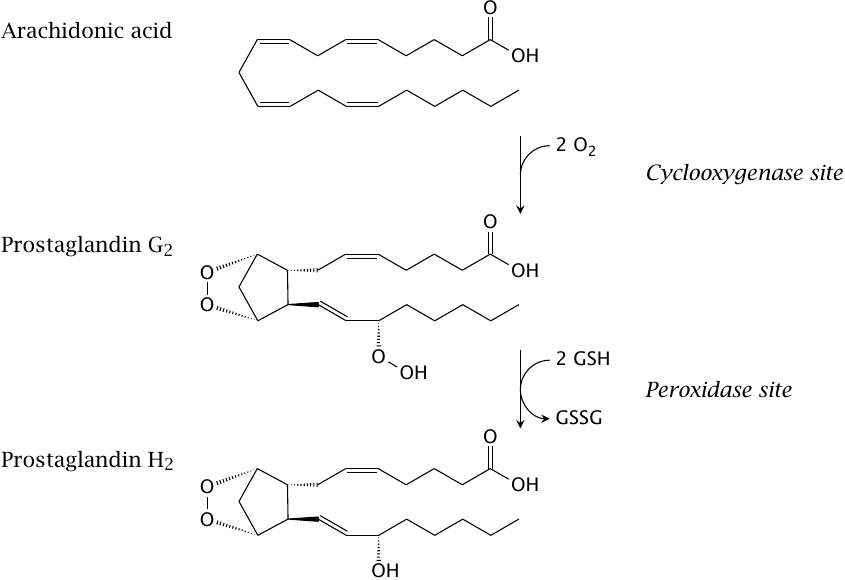
The cyclooxygenase reaction occurs in two stages, which are catalyzed in two separate active sites. In the cyclooxygenase site, arachidonic acid reacts with two molecules of O2 to acquire an endoperoxide and a hydroperoxide group; this yields the intermediate prostaglandin G2. The hydroperoxide is then reduced to a simple hydroxyl group in the peroxidase site, which gives prostaglandin H2.
While the two active sites are quite close in space, there is no direct pathway for the substrate to migrate from one to the other. Prostaglandin G2 must therefore dissociate from the cyclooxygenase site and then rebind to the peroxidase site of the same or another enzyme molecule.
| 9.3.1 |
Reactions in the cyclooxygenase site |
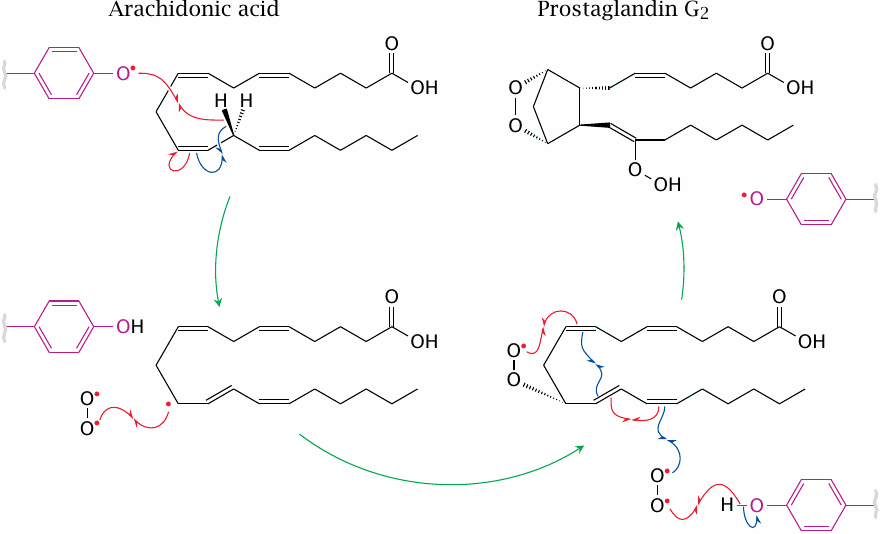
The key feature of the cyclooxygenase active site is a tyrosine residue (Tyr385 in Cox-1; highlighted in purple) which, in order to start the reaction, must be a radical. This radical abstracts a single hydrogen from arachidonic acid, which thereby itself turns into a radical. The radical electron then migrates and combines with an O2 π-diradical.
A cascade of further radical reactions and bond migrations lead to cyclization and to radical combination with a second O2 molecule, which then reclaims the hydrogen from the tyrosyl residue and thus restores the enzyme to its active state. The product, prostaglandin G2, then leaves the cyclooxygenase site to find a peroxidase site.
| 9.3.2 |
Reduction of prostaglandin G2 to prostaglandin H2 |
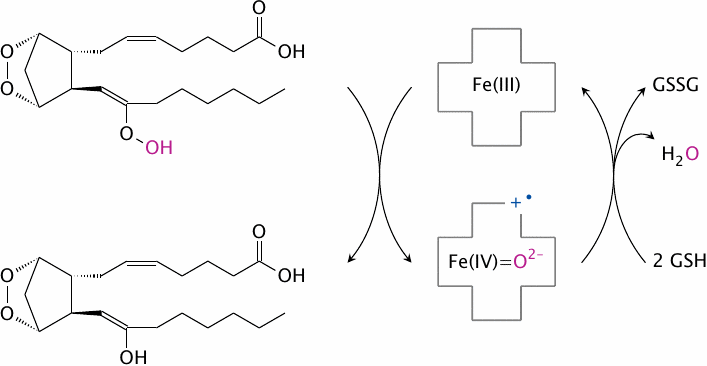
The peroxidase site contains heme as a prosthetic group. This heme “advances” one electron to each of the oxygen atoms in the peroxy group of the substrate, reducing their formal charges from −1 to −2 and retaining one of them. The heme itself is reduced in turn using two molecules of glutathione.
| 9.3.3 |
Interaction between the cyclooxygenase and peroxidase sites |
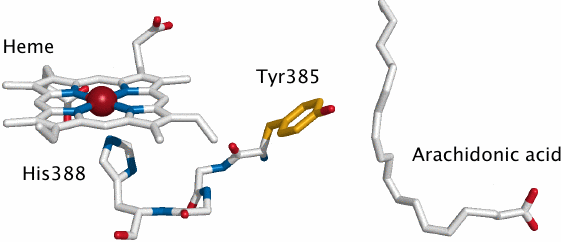
The two active sites in the cyclooxygenase molecule are close in space, and an interaction between them is required to convert the tyrosine in the cyclooxygenase site to its active radical form. In this interaction, an electron is abstracted from the reduced form of the tyrosine and consumed in the peroxidase reaction, replacing one of the electrons derived from glutathione in the regular peroxidase reaction.
In the picture, arachidonic acid is shown to indicate the location of the cyclooxygenase site; Tyr 385 is the catalytic tyrosine residue in that site. It is connected to the heme in the peroxidase site via a short stretch of three amino acid residues, which reportedly acts as the electron conduit between it and the heme.
| 9.3.4 |
Priming of the active site tyrosine radical, and the action mode of acetaminophen |
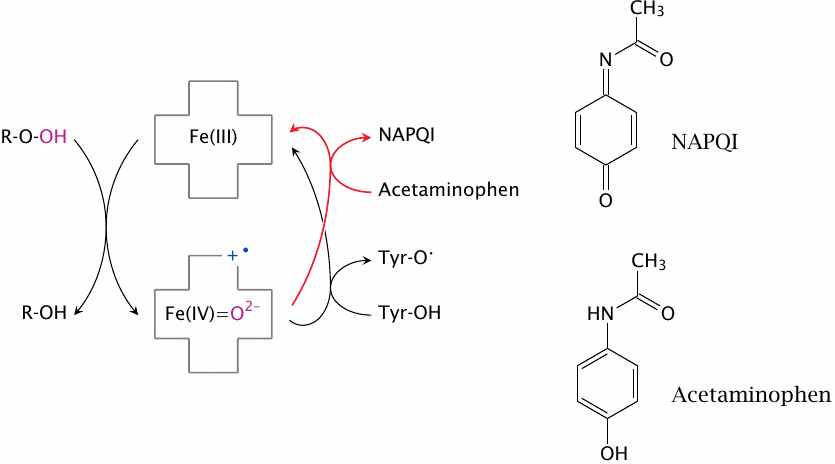
The activation of tyrosine 385 starts with the oxidation of heme by prostaglandin G2 or another hydroperoxide (ROOH). The oxidized heme then obtains one electron from tyrosine 385 via the conduit shown in the preceding slide, thereby activating it; another electron is presumably obtained from glutathione (not shown). Acetaminophen intercepts the activation of tyrosine 385 by reducing heme itself.
The inhibition of Cox by acetaminophen is countered by high cellular levels of hydroperoxides. Such high concentrations exist in leukocytes, which produce hydrogen peroxide and other reactive oxygen species as antimicrobial effectors (see slide 8.6). Therefore, while acetaminophen has good activity in neurons or sensory cells and therefore suppresses pain and fever, it has little effect on the synthesis of proinflammatory prostaglandins in leukocytes. Better antiinflammatory activity is observed with drugs that inhibit the enzyme in a redox-independent fashion.
| 9.4 |
Noncovalent cyclooxygenase inhibitors |
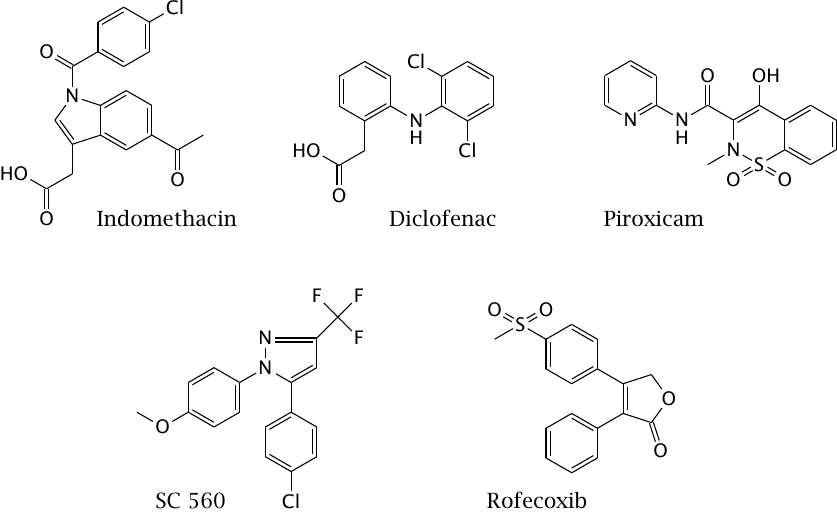
The drugs shown in this slide all inhibit the cyclooxygenase site of the enzyme. The three drugs in the top row all inhibit both Cox-1 and Cox-2, whereas SC560 inhibits only Cox-1, and rofecoxib only Cox-2. All drugs bind to the enzyme non-covalently.
Cox-2 is prominently expressed by inflammatory cells, and its selective inhibition was once believed to avoid potentially serious side effects that may arise from the simultaneous blockade of physiological “housekeeping” functions, which were credited to Cox-1. However, the opposite has turned out to be true: a significant increase in the frequency of myocardial infarctions under treatment with rofecoxib and other selective Cox-2 inhibitors has prompted their withdrawal from the market. Indeed, experiments with genetic knockouts of Cox-1 [73] and Cox-2 [74] had thrown the assumed “good cop, bad cop” story into question before the Cox-2 inhibitors had even been introduced into the clinic; it is a bit perplexing that after these reports those efforts continued unabated.
| 9.4.1 |
Conformation of arachidonic acid and of diclofenac in the active site |
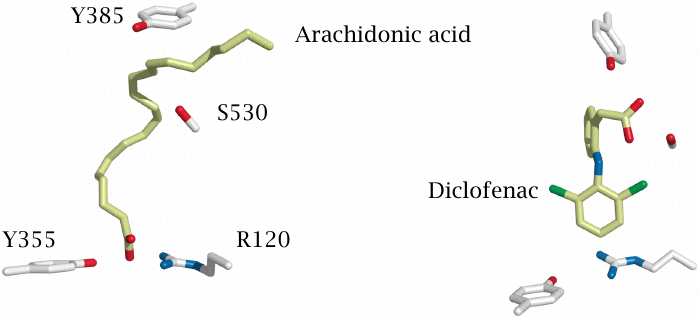
Like arachidonic acid, both indomethacin and diclofenac have a carboxylic acid group, and one might reasonably surmise that all three carboxylates interact with the same amino acid residues in the active site. However, while this is true for arachidonic acid and indomethacin, crystallography shows that diclofenac sits in the active site upside down.
The carboxyl groups of arachidonic acid (left) and of indomethacin (not shown) bind to tyrosine 355 and arginine 120. In contrast, the carboxyl group of diclofenac (right) is hydrogen-bonded to serine 530 and to the catalytic tyrosine 385. Structures rendered from 1diy.pdb and 1pxx.pdb.
| 9.4.2 |
Cox inhibitors and Cox mutants |
| Inhibitor | Relative IC50 | ||
| R120A | Y355F | S530A | |
| Indomethacin | > 240 | > 240 | 1.1 |
| Diclofenac | 3.3 | 1.8 | > 650 |
| Piroxicam | > 109 | > 109 | > 109 |
The above crystallographic findings agree with those from site-directed mutagenesis experiments. Arginine 120, tyrosine 255, or serine 530 were replaced with alanine or phenylalanine, respectively, whose side chains cannot engage in hydrogen bonding. An increase in the inhibitor concentration required for 50% inhibition (IC50) relative to the wild type enzyme indicates that the mutant has become less susceptible to the inhibitor.
Mutants that lacked arginine 120 or tyrosine 355 were virtually resistant to indomethacin. In contrast, their inhibition by diclofenac was hardly affected by these changes, but was very sensitive to replacement of residue serine 530. Interestingly, piroxicam was susceptible to all three changes, indicating that avid binding of this inhibitor involves all three wild type residues. Data from [75].
| 9.5 |
Acetylsalicylic acid is a covalent Cox inhibitor |
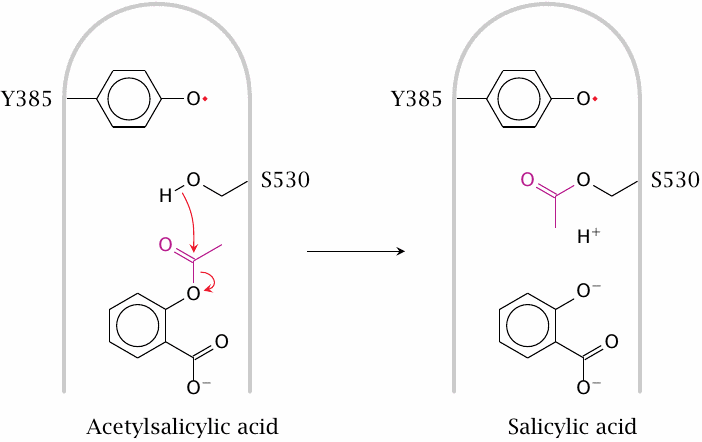
Acetylsalicylic acid covalently modifies the side chain of serine 530 in the active site of Cox-1. This prevents access of arachidonic acid to the catalytic tyrosine and therefore inactivates the enzyme. Note that, in contrast to cholinesterase for example, the serine residue itself has no catalytic function; the interference of its modification with Cox activity is purely due to steric obstruction.
The drug also acetylates the homologous serine residue within Cox-2. The active site of Cox-2 has a larger interior volume, which means that access of arachidonic acid to the catalytic tyrosine is not completely prevented, and the reaction not completely suppressed. However, the reaction is “derailed”, such that the modified enzyme no longer makes prostaglandin H2 but instead now converts arachidonic acid to 15-R-HETE, which through additional enzymatic steps gives rise to epi-lipoxin A4. Like the physiological mediator lipoxin A4 (see slide 9.2.1), this compound has antiinflammatory activity and likely contributes to the therapeutic action of acetylsalicylic acid [76].
| 9.5.1 |
Rationale of low-dose acetylsalicylic acid treatment |
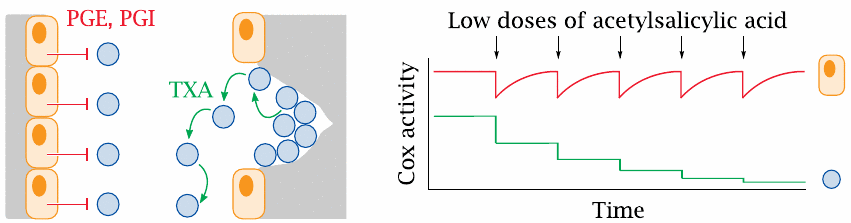
Patients with advanced atherosclerosis are at risk of myocardial infarction or stroke, which develop when atherosclerotic lesions in the vascular endothelium trigger the formation of blood clots (see slide 10.6). Thrombocyte activation and aggregation is a key factor of blood clot formation; therefore, drugs that inhibit thrombocyte aggregation69 play an important part in the preventive treatment of such patients. An early form of such treatment was low-dose acetylsalicylic acid. The rationale for this treatment is not obvious but rather subtle.
Eicosanoids have a dual role in thrombocyte aggregation (left). The vascular endothelium inhibits aggregation through sustained secretion of PGE and PGI. Where the endothelium is damaged, exposure of collagen sets off thrombocyte adhesion and aggregation, which is amplified through the secretion of thromboxanes by the platelets themselves. Both the thrombocyte-activating thromboxanes and the inhibitory prostaglandins E and I are synthesized downstream of prostaglandin H2. Acetylsalicylic acid inhibits the synthesis of this common precursor by cyclooxygenase; why should this result in a net inhibition of aggregation?
The answer lies in the different lifetimes of the enzyme molecules in endothelial cells and in thrombocytes, respectively (right). Endothelial cells are nucleated and can, after each dosage of the drug, replace covalently inactivated cyclooxygenase molecules with newly synthesized ones. In contrast, thrombocytes lack nuclei and protein synthesis, and therefore cannot replace the modified enzyme molecules; the effect of repeated doses will thus be cumulative. Over time, cyclooxygenase activity will therefore be diminished much more in thrombocytes than in endothelial cells.
| 9.5.2 |
Cox inhibition can promote the synthesis of leukotrienes |
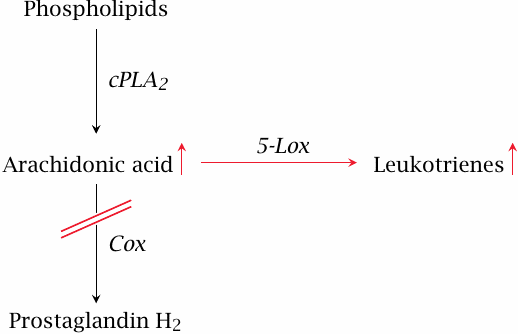
As we have seen in slide 9.1.2, the cyclooxygenase and lipoxygenase enzymes draw from the same pool of arachidonic acid substrate. Therefore, if cyclooxygenase is inhibited, more substrate may feed into the synthesis of leukotrienes which, like several prostaglandins, have proinflammatory activity. A well-known clinical consequence of this effect is the exacerbation of bronchial asthma in patients who take acetylsalicylic acid.
The production of surplus leukotrienes illustrates that inhibition of Cox as an antiinflammatory strategy has its limitations, and that more selective strategies for controlling eicosanoid action are desirable.
| 9.6 |
Inhibitors that act downstream of Prostaglandin H2 |
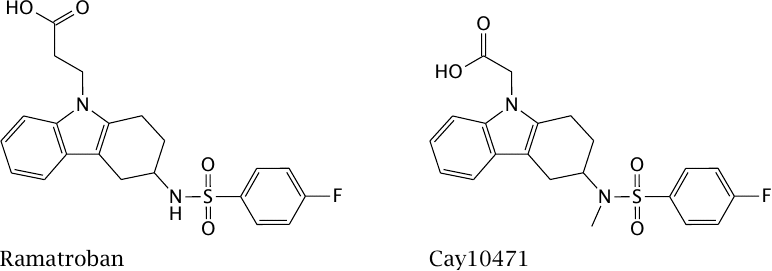
Thromboxane A synthase is the enzyme that converts prostaglandin H2 to thromboxane A2 (see slide 9.2). The enzyme, as well as the receptor for thromboxane A2, are inhibited by the drug ramatroban.
Prostaglandin D is involved both in inflammation and pain. The experimental drug Cay 10471, which is structurally similar to ramatroban, inhibits prostaglandin D receptors.
| 9.7 |
Endocannabinoids |
- Arachidonate-containing, membrane-derived mediators
- Synthesis on demand, activated by calcium
- Mediate negative synaptic feedback
- CB1 receptors involved in pain perception both in the peripheral and the central nervous system
- CB2 receptors found on immune cells
We have seen before that postsynaptic cells exercise negative feedback on presynaptic cells (see slide 6.9.1). The mediators that perform this role are endocannabinoids. Two major endocannabinoids are arachidonylethanolamine (AEA, anandamide) and arachidonylglycerol (2-AG). These mediators act on cannabinoid receptors, which are also the targets of the major active ingredients of cannabis.
Cannabinoid receptors are GPCRs and have two subtypes. CB1 receptors dominate in the nervous system and are the major drug target. CB2 receptors modulate immune cell function, but this observation has not yet translated into pharmacotherapy.
| 9.7.1 |
Feedback inhibition of synaptic transmission by endocannabinoids |
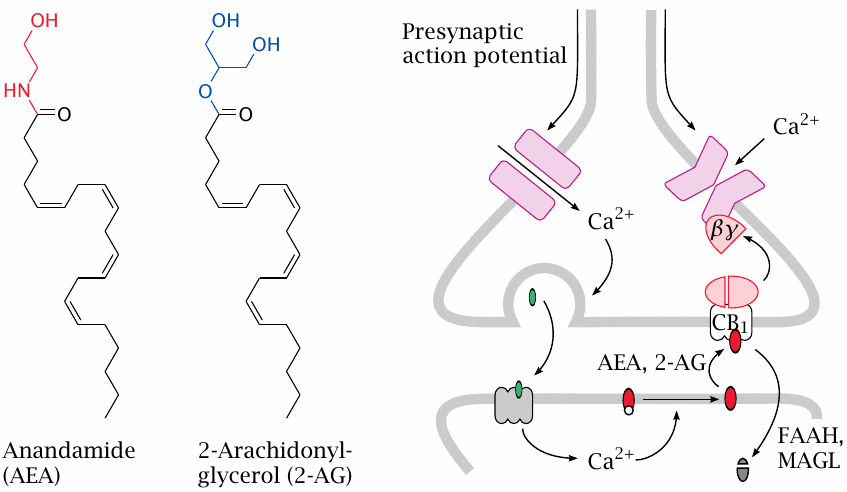
Anandamide and arachidonylglycerol both contain arachidonic acid attached to a polar moiety that is biosynthetically derived from a phospholipid headgroup. Both mediators are synthesized in the postsynaptic cell in response to calcium signals; as we have seen, several neurotransmitter receptors will either directly or indirectly raise calcium levels.
The mediators are released by the postsynaptic cell into the synaptic cleft. Stimulation of presynaptic CB1 receptors activates G proteins; the released Gβγ dimers inhibit cognate Ca++ channels and also activate K+ channels, thereby reducing membrane excitability and transmitter release. The mediators are inactivated by cellular uptake and subsequent degradation by lipases (fatty acyl amide hydrolase, FAAH; monoacylglycerol lipase, MAGL; see slide 9.7.3).
| 9.7.2 |
Biosynthesis of endocannabinoids |
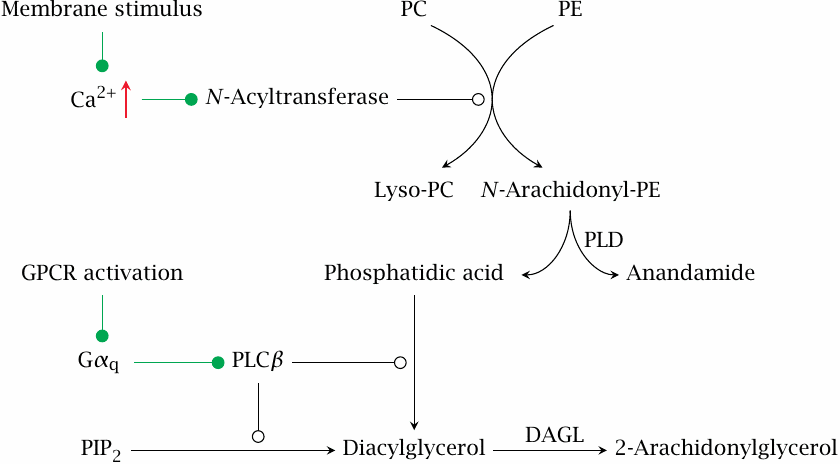
This slide shows the biosynthetic pathways that produce anandamide (N-arachidonylethanolamine) and 2-arachidonylglycerol (2-AG).
Increased Ca++ levels activate an N-acyltransferase that transfers arachidonic acid from another phospholipid molecule to the amine in the headgroup of phosphatidylethanolamine (PE). Anandamide is then released by phospholipase D (PLD). 2-AG is produced by diacylglycerol lipase (DAGL) downstream of the phospholipase C pathway.
| 9.7.3 |
Degradation of endocannabinoids |
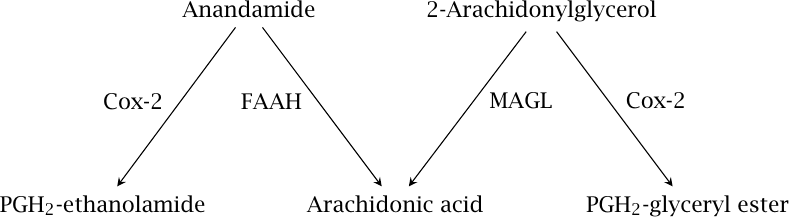
Anandamide is hydrolyzed by fatty acid amide hydrolase (FAAH) and 2-arachidonylglycerol by monoacylglyerol lipase (MAGL). Both mediators can also be metabolized by cyclooxygenase 2 to produce prostamides, which have additional signaling roles that are not yet well characterized.
| 9.7.4 |
Drugs that interact with the endocannabinoid system |
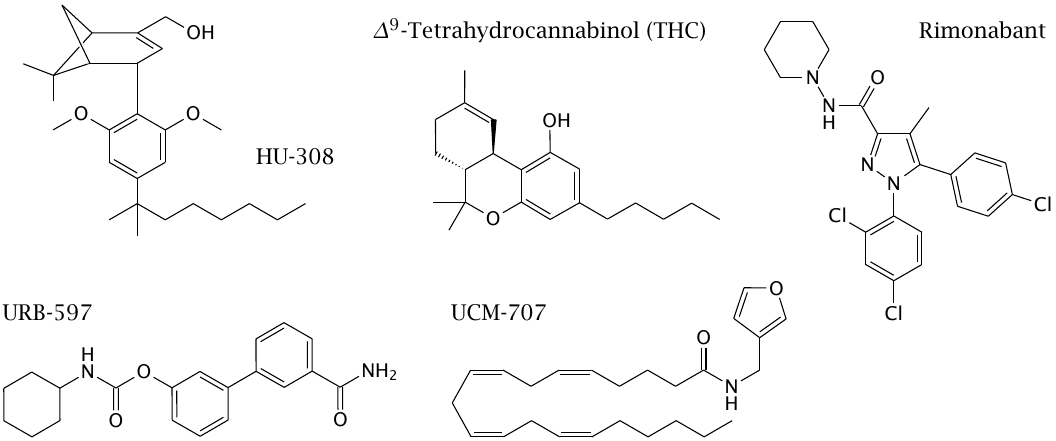
Δ9-tetrahydrocannabinol (THC) is the main active ingredient of cannabis and stimulates both CB1 and CB2 receptors. HU-308 is a CB2-selective agonist. URB-597 inhibits fatty acyl amide hydrolase. UCM-707 is usually described as an inhibitor of cellular uptake of endocannabinoids. However, it is not clear at present that there indeed exists a specific transporter for this reuptake; this compound, too, might inhibit the hydrolase. The functional effect of both inhibitors is to amplify endocannabinoid signaling.
Rimonabant is an inverse agonist at CB1 receptors. It has been used to reduce appetite in order to treat obesity, but was withdrawn because depression was a common side effect.