| 14 |
Drug delivery |
| 14.1 |
Introduction |
As we have seen in chapter 3, some drugs cannot get to their targets easily or even at all. In some cases, the best strategy is to use another drug altogether; however, in other cases, we can take measures to improve the uptake and retention of drugs, or to target them to specific sites of action in order to improve effectiveness and reduce toxicity. Such auxiliary techniques are collectively referred to as drug delivery. This chapter discusses a number of instructive examples.
| 14.2 |
Modifying drug molecules to improve intestinal uptake and distribution |
We have already considered this topic earlier (see slide 3.4.5). Here, we will expand on it with several more examples of techniques and applications.
| 14.2.1 |
Protecting drugs from gastric acid through prodrug formation |
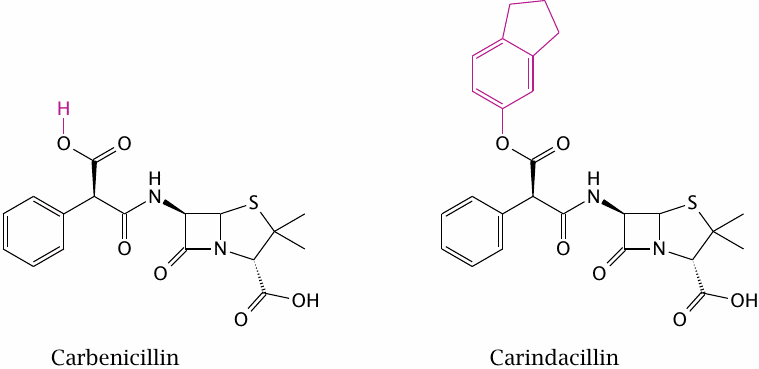
Carindacillin is an orally applicable prodrug of carbenicillin, a penicillin derivative with good activity against various Gram-negative bacteria (but one which is no longer commonly used). Esterification with indanol (highlighted) improves acid resistance. In addition, it renders the prodrug more hydrophobic than the parent compound and promotes its uptake from the intestine.
| 14.2.2 |
Optimizing a drug structure for bilayer permeation |
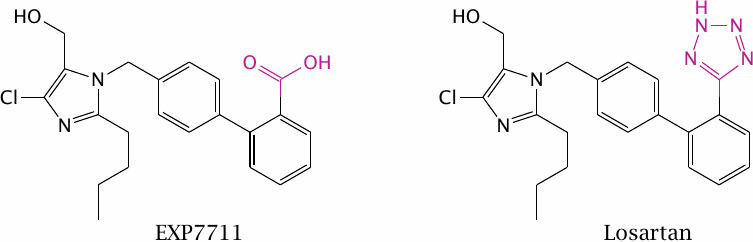
EXP7711 is an angiotensin receptor blocker with antihypertensive activity. The acidic carboxylate group in EXP7711 inhibits intestinal absorption, yet the negative charge in this position is necessary for tight receptor binding in order to achieve a high IC50.
Like carboxylate, tetrazole is acidic and has a planar structure, but at the same time it is ten times more lipophilic. Replacement of the carboxylate in EXP7711 with tetrazole gives losartan, which retains inhibitory activity on the angiotensin receptor (see slide 1.2.5) yet is absorbed from the intestine much more readily.
| 14.2.3 |
Trapping an estradiol prodrug inside the brain |
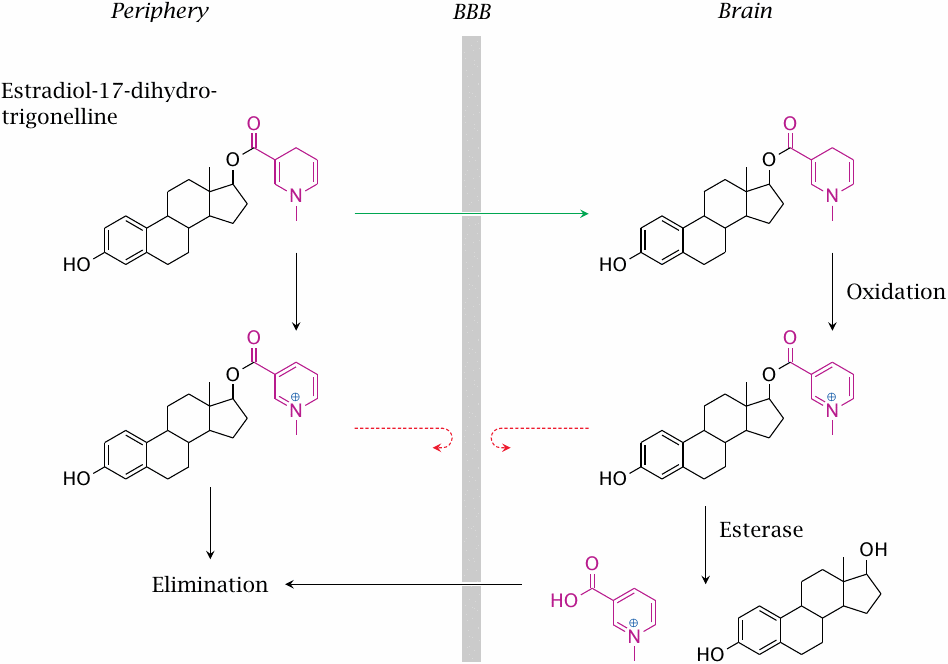
The 1,4-dihydro-N-methylnicotinic acid (trigonelline) ester of estradiol can enter the brain by diffusion. Oxidation of the trigonelline moiety occurs both in the periphery and the brain. The charge introduced by oxidation will accelerate elimination in the periphery but inhibit elimination from the brain, since the charged molecule is no longer able to cross the BBB. Estradiol is slowly released from the trapped prodrug by esterases.
| 14.2.4 |
Succinylsulfathiazole, a prodrug designed for reduced absorption |
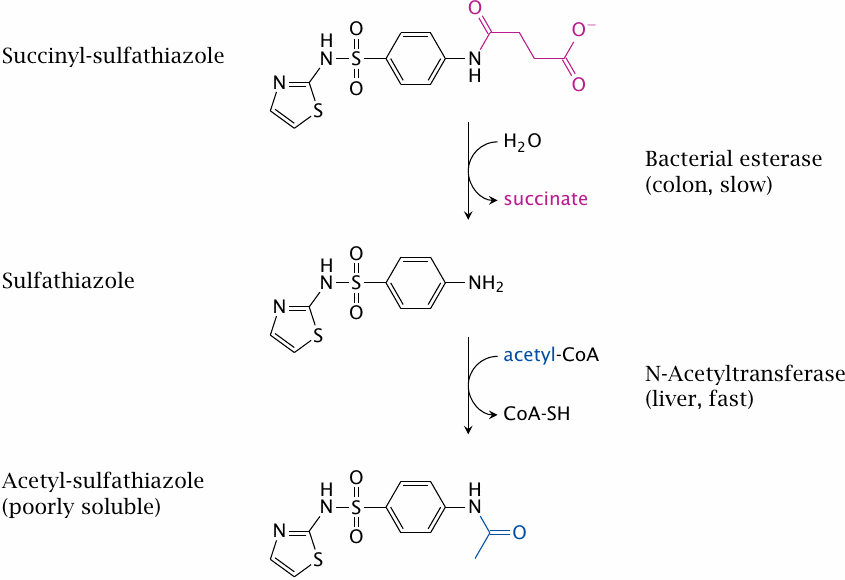
Sulfathiazole is a sulfonamide like sulfamidochrysoidine (see slide 1.3.3) and shares its mode of action. Sulfathiazole is metabolized through N-acetylation. The acetylated compound is poorly soluble, which may cause it to precipitate inside the kidney tubules; this may be lethal.
Succinylsulfathiazole is less toxic because it is ionized and thus not taken up efficiently. Once the drug reaches the large intestine, it is slowly hydrolyzed by bacterial esterases. The low rate of hydrolysis means that its uptake will also be slow and protracted; systemic concentrations of sulfathiazole and the acetylated metabolite will remain low throughout, and renal complications will be avoided.
While succinylsulfathiazole illustrates an approach to mitigating drug toxicity that is interesting in principle, in practice we prefer to use other sulfonamides that are not encumbered with this kind of potential problems in the first place.
| 14.3 |
More on dopamine and its prodrugs |
| 14.3.1 |
Inhibition of DOPA decarboxylase in the periphery improves l-DOPA uptake into the brain |
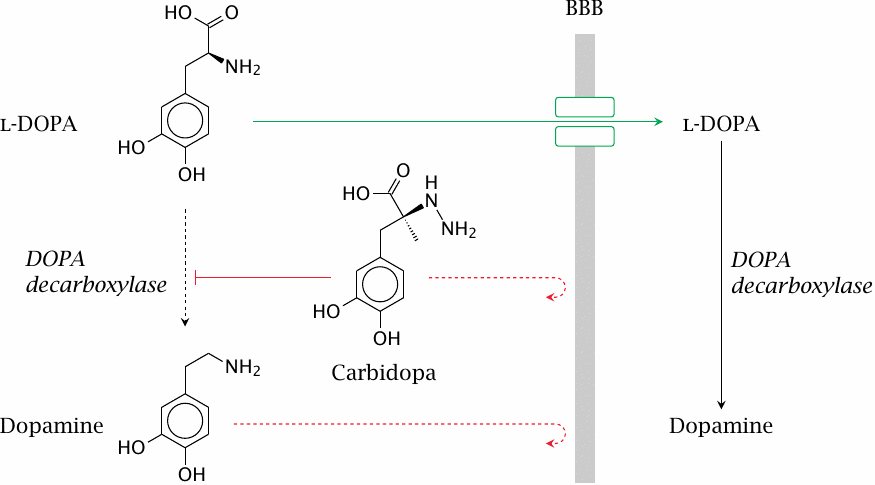
As discussed earlier (slide 3.5.5), dopamine does not cross the blood brain barrier (BBB); therefore, in the treatment of Parkinson’s disease, its metabolic precursor l-DOPA is used as a prodrug. l-DOPA crosses the BBB by active transport and is converted to dopamine in the brain.
DOPA decarboxylase, the enzyme that converts l-DOPA to dopamine, is active both in the periphery and the CNS. Accordingly, if l-DOPA is applied alone, most of it undergoes decarboxylation in the periphery. The dopamine produced there cannot enter the brain but may instead be converted to norepinephrine or epinephrine and so cause side effects on blood pressure and glucose metabolism.
The premature conversion of l-DOPA can be prevented by combining l-DOPA with carbidopa or benserazide, which inhibit DOPA decarboxylase in the periphery. Like dopamine, these compounds do not cross the BBB and thus do not interfere with dopamine metabolism in the brain. Combination with such inhibitors permits the dosage of l-DOPA to be reduced by ~75% and very significantly reduces side effects.
| 14.3.2 |
Gludopa, a prodrug for selective release of dopamine in the kidneys |
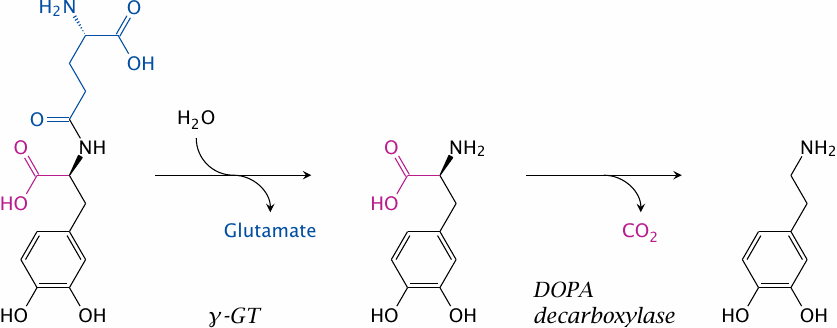
Dopamine receptors in the kidney vasculature improve kidney perfusion and function, and their selective stimulation may be clinically desirable. A prodrug to target dopamine preferentially to the kidneys is gludopa (γ-glutamyl-l-DOPA).
The kidneys (as well as the liver) contain l-γ-glutamyl-transpeptidase (γGT) at high levels of activity. This enzyme transfers γ-glutamyl residues between different substrates, but can also simply hydrolyze them. Hydrolytic removal of γ-glutamate from gludopa in the kidneys releases l-DOPA, which is again converted to dopamine and stimulates the local dopamine receptors.
| 14.4 |
Using carriers and vehicles to improve drug delivery |
Aside from modifying the drug molecules themselves, the distribution or organ targeting of drugs can sometimes be improved by suitably packaging them before application.
| 14.4.1 |
Protecting drugs from gastric acid through acrylate copolymer coating |
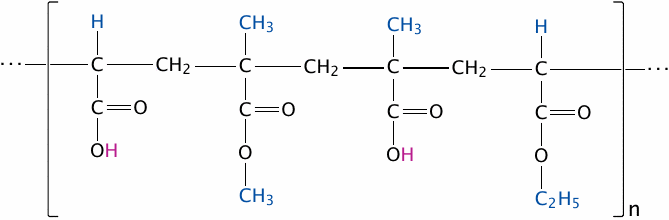
Some drug molecules contain acid-labile bonds such as carboxylic acid esters. These drugs must be protected from destruction by the fairly concentrated hydrochloric acid in the stomach. One way to achieve this protection is through micro-encapsulation in polymers that remain stable in an acidic milieu but dissolve once they reach the slightly alkaline environment within the small intestine. This technique is commonly referred to as enteric coating.
Widely used for this purpose are acrylate copolymers, often referred to using their trade name (Eudragit®). The acidic pH inside the stomach keeps the polymers’ carboxylic acid groups protonated, and the whole particles insoluble. Upon entering the small intestine, the carboxylic acid groups dissociate, and the ester bonds are cleaved. The polymers disperse and dissolve, releasing the cargo drugs enclosed within.
The slide shows one specific example, namely Eudragit® L 100-55; the substituents highlighted in blue differ in other commercial grades of this product (see [132] for details).
| 14.4.2 |
Site-selective delivery of BCNU |
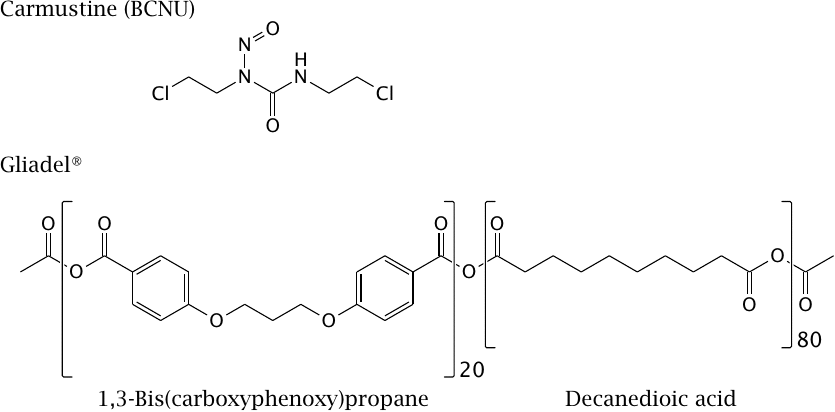
Bis-chloroethyl-nitrosurea is a bifunctional alkylating agent that is used in the treatment of cancer (see slide 12.6.18). The drug can cross the BBB in principle, but it is highly reactive and therefore consumed within a short range of penetration into the brain tissue, mostly before reaching the tumor. Therefore, systemic application for treating brain tumors is problematic.
For localized, sustained delivery in treating brain tumors, the drug is embedded in a polyanhydride polymer, and the product is shaped into wafers that go by the trade name Gliadel®. These are placed into the tumor resection cavity during tumor surgery.
The ratio of carboxyphenoxypropane and sebacic acid building blocks in the polymer can be varied to control the rate of its hydrolysis and accordingly of drug release from the polymer matrix.
| 14.4.3 |
Cyclodextrins: Structure and use in drug delivery |
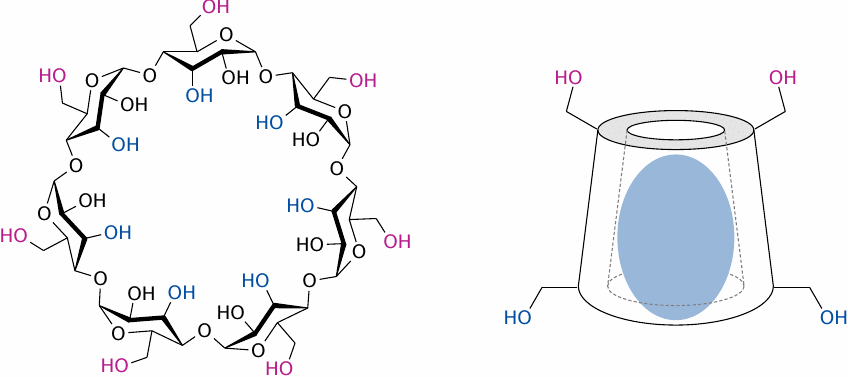
Cyclodextrins are naturally occurring, circular glucose polymers. In aqueous solution, they adopt a conformation that encloses a central cavity, which can accommodate a hydrophobic guest molecule. The variants α-, β- and γ-cyclodextrin contain 6, 7 and 8 molecules of glucose, respectively. β-Cyclodextrin is most commonly used, but the others are of use with smaller or larger drug molecules.
Cyclodextrins are small and polar enough to be renally eliminated, and their degradation will produce simply glucose; therefore, they are well tolerated and suitable for both oral and parenteral application. Piroxicam (see slide 9.4.2) and prostaglandin E1 are examples of drug molecules that are applied as cyclodextrin complexes.
Left: Structure of β-cyclodextrin. Right: Schematic of a cyclodextrin molecule encasing a hydrophobic drug molecule.
| 14.4.4 |
Solubilization of hydrophobic drugs with surfactants |
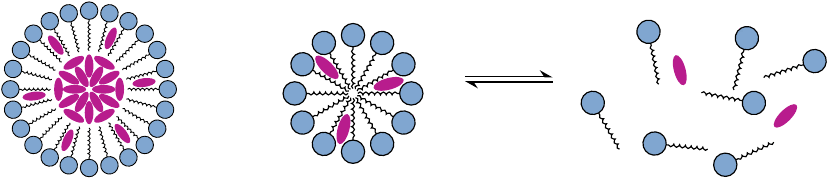
Surfactants, or detergents, are amphiphilic molecules that in aqueous solution aggregate into micelles. These micelles can accommodate hydrophobic drug molecules as aggregates (left) or as individual molecules (center).
Most surfactants rapidly equilibrate between the micellar and the monomeric state. When the surfactant is diluted to below its critical micellar concentration, the micelles will rapidly dissipate, and the drugs contained within will be released (right). The antifungal drug amphotericin B (see slide 11.8.1) used to be delivered in this manner, using deoxycholic acid as the detergent. However, as we will see below, liposomal delivery of this drug reduces toxicity and as now preferred.
| 14.4.5 |
Liposomes as drug delivery vehicles |
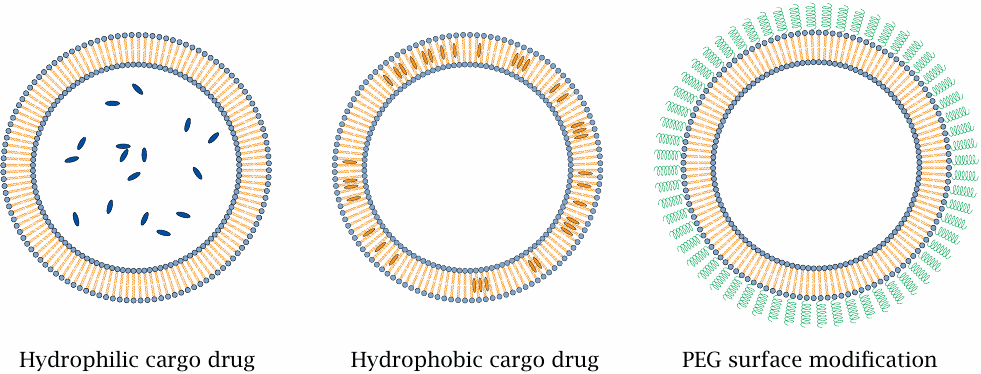
Liposomes are vesicles consisting of natural or synthetic phospholipids. They can vary widely in size, but the ones used for drug delivery are most often between 50 and 200 nm in diameter. Hydrophilic cargo drugs will be enclosed in the lumen (left), whereas hydrophobic ones will reside in the bilayer itself (center).
The phospholipids that constitute the liposomes have negligible water solubility; therefore, unlike detergent micelles, they will not simply disperse after intravenous application and dilution. However, their lifetime may still be cut short due to encounters with proteins of the complement system in the blood plasma, which may disrupt their membranes and mediate their removal from the circulation by phagocytosis. Disruption by complement proteins can be prevented through surface modification with polyethyleneglycol (PEG), which will greatly increase their stability and dwell time in the circulation (right).
| 14.4.6 |
Liposomal vs. deoxycholate-solubilized amphotericin B in a mouse infection model |
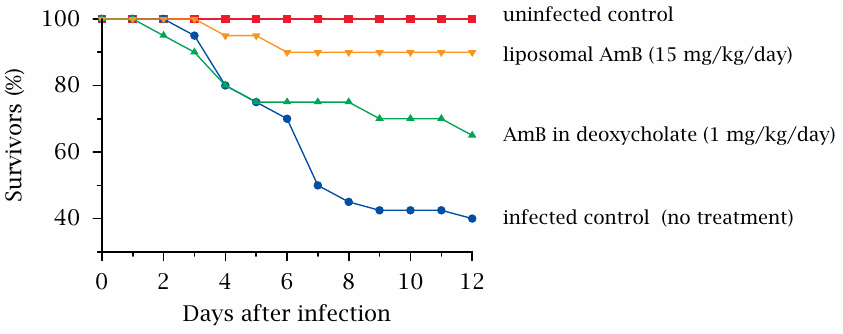
Amphotericin B is an antifungal antibiotic that binds to ergosterol in the fungal cell membrane, which it then renders permeable toward cations (see slide 11.8.1). The molecule is fairly hydrophobic and therefore must be solubilized in some way. One widely used preparation contains the surfactant deoxycholate, but liposomal preparations are now frequently used because of their greater therapeutic index.
The lower toxicity of liposomal amphotericin B (AmB) is illustrated here in a mouse model of a fungal infection. In preliminary experiments, the highest tolerable dosage for both the deoxycholate-based and the liposomal AmB preparations was established; a fifteen-fold higher dosage was tolerated when the drug was bound to liposomes. When these respective dosages were applied to mice that had been infected with the rather nasty fungal pathogen Rhizopus oryzae, the liposomal preparation came out ahead. Figure prepared from original data in [133].
Another interesting study explored the use of the related polyene antibiotic nystatin as a liposomal preparation. This drug had traditionally been considered too toxic for systemic application; however, when delivered using liposomes, the drug was tolerated by mice and had equal or better activity than amphotericin B against experimental infection with Aspergillus fumigatus [134].
| 14.4.7 |
Liposomes and the Enhanced Permeability and Retention (EPR) effect |
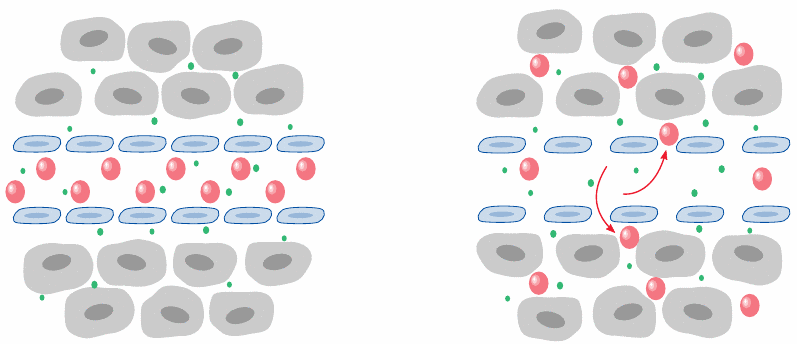
Liposomes can also be used for targeted—or preferential—delivery of cytotoxic drugs to cancer tissue. In cancer, as well as in inflammation, the capillary barriers become leaky, and plasma proteins that are normally confined to the circulation can disperse in the interstitial space.110
The leakiness also applies to liposomes up to approximately 200 nm in diameter, which will therefore preferentially accumulate in cancers and cancer metastases. PEG-modified liposomes loaded with the anticancer drug doxorubicin (see slide 12.6.19) have been FDA-approved for tumor therapy [135].
| 14.5 |
Antibodies as drug-targeting devices |
Antibodies have exquisite affinity and selectivity for their targets (antigens) and can be used to direct drugs to specific tissues and cell. In some cases, the antibodies themselves can act as drugs; an example is “trastuzumab”, the monoclonal antibody directed against the tumor-related growth factor receptor Her2/neu. Alternatively, the antibodies may be labeled with a radioactive isotope or coupled with a cytotoxic drug such as calicheamicin (see below), or they may be attached to the surface of drug-containing liposomes.
| 14.5.1 |
Humanized antibodies |
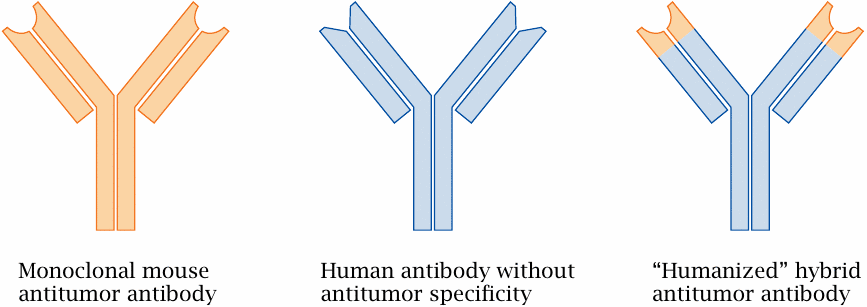
Monoclonal antibodies are conventionally derived from mice and thus immunogenic to humans, which means that patients receiving repeated applications may develop anti-antibodies that bind and neutralize them. The immunogenicity of mouse antibodies can be substantially reduced by creating hybrid molecules that retain only the variable, antigen-specific moieties from the mouse antibodies but derive the invariant remainder of the molecule from a human antibody.
| 14.5.2 |
A calicheamicin-antibody conjugate |
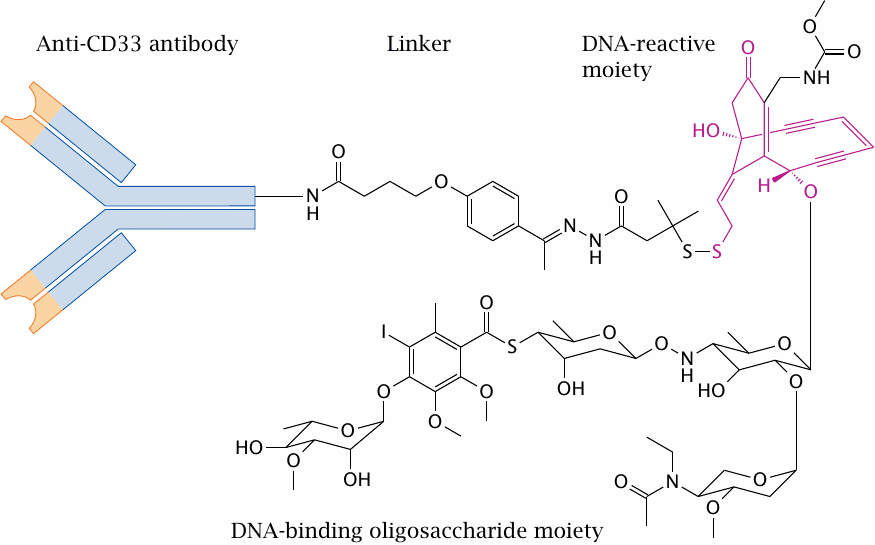
This conjugate—poetically named “gemtuzumab ozogamicin”—contains a humanized IgG monoclonal antibody (hP 67.6), which binds to CD33, a surface antigen found on leukemic cells in acute myeloic leukemia (AML). A derivative of the cytotoxic antibiotic calicheamicin γ is linked to the antibody by (4-acetylphenoxy)butanoic acid. Two sugar residues within the oligosaccharide moiety of the antibiotic are responsible for binding of the minor groove of DNA. Several calicheamicin molecules may be coupled to one antibody. The covalent reaction of calicheamicin with DNA is explained below.
The conjugate was FDA-approved for a brief period of time but has since been withdrawn due to unacceptable toxicity.
| 14.5.3 |
Activation of calicheamicins |
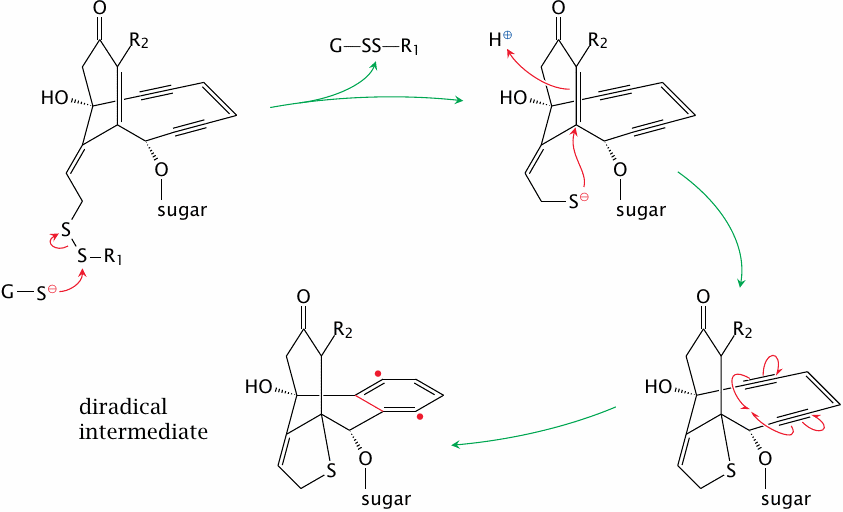
Calicheamicins are endiyne antibiotics, that is, they possess one alkene bond between two conjugated alkyne bonds. They also have a trisulfide group, which in the antibody conjugate discussed above is replaced by a disulfide.
Once the drug enters the cell, an intracellular nucleophile, usually glutathione, attacks the di- or trisulfide. Subsequent conjugate addition of the enone and Bergman rearrangement generate a reactive diradical intermediate, which reacts with DNA and induces DNA double strand breaks (see next slide).
| 14.5.4 |
DNA cleavage by activated calicheamicins |
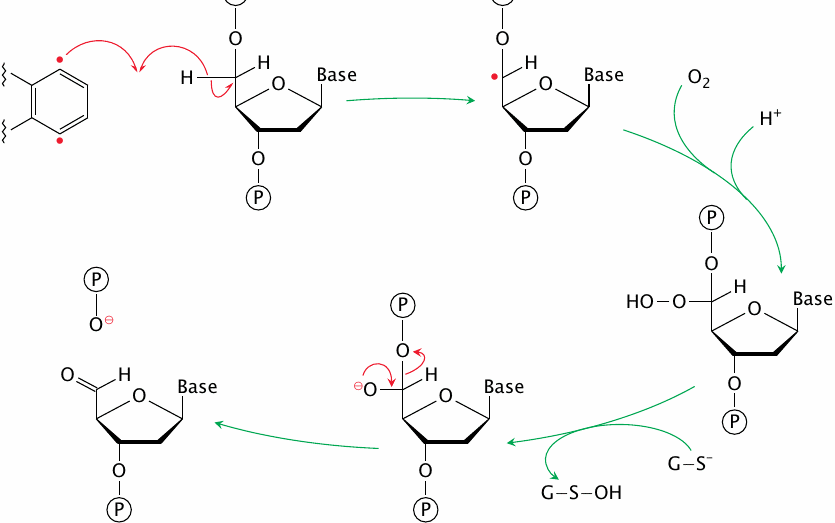
This slide shows the reaction of one of the two radicals in activated calicheamicin with a single DNA strand. Abstraction of a hydrogen atom from deoxyribose produces a carbon radical, which reacts with molecular oxygen to form a peroxide. The latter is reduced by glutathione to an alkoxide, which then cleaves the adjacent phosphoester bond.
Hydrogen abstraction and bond cleavage can occur at other positions of the deoxyribose moiety and result in other outcomes, including abasic sites. A common outcome seems to be cleavage of one DNA strand, combined with an abasic site on the other [136].
| 14.6 |
Delivery of insulin |
Most body cells require insulin to facilitate uptake of glucose from the blood, and while the plasma insulin level rises and falls in keeping with blood glucose levels, the hormone must be continuously present in the blood in order to maintain metabolism in good working order. In diabetes mellitus, insulin is lacking or insufficient and must be substituted (see slide 10.5).
The plasma half-life of insulin molecules, once secreted, is only about ~15 minutes; the hormone is depleted both by peptidases and by receptor binding. Therefore, plasma levels have to be sustained by continued secretion from the pancreas. In insulin replacement treatment of diabetics, various techniques are used in order to achieve a sustained level of blood plasma insulin. A modern development are insulin pumps that deliver insulin continuously; still more commonly used, however, are techniques that achieve a protracted uptake of subcutaneously injected insulin into the circulation.
| 14.6.1 |
Aggregation of insulin delays its uptake into the circulation |
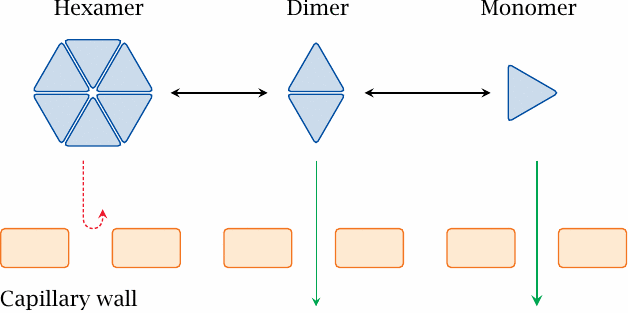
If insulin is injected intravenously, it will rapidly be inactivated through cleavage by peptidases. To achieve a more sustained activity, insulin is injected subcutaneously, that is, into the interstitial space.
Insulin exists in different states of association. Zinc ions and high insulin concentrations promote the formation of hexamers, which are too large to permeate across the capillary walls. The aggregation equilibrium can be shifted either way by point mutations or chemical modifications, which is exploited in the preparation of both fast- and slow-acting insulins.
Slow-acting insulins are optimized for subcutaneous application 2–3 times per day; each of the injected aliquots must then release insulin slowly over a time period of many hours. In contrast, fast-acting insulins are optimized for more frequent injections, or for use with insulin pumps, which infuse insulin continuously and at a rate that is adjusted according to frequent measurements of blood glucose.
| 14.6.2 |
Structure of the insulin hexamer |
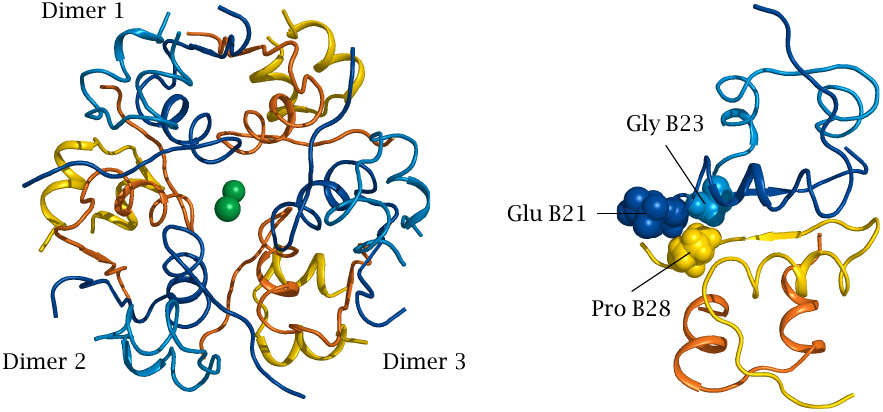
The insulin hexamer is composed of three dimers and stabilized by two centrally placed zinc ions. In each dimer, proline 28 of one B chain interacts with glutamate 21 and glycine 23 of the opposite B chain. In the recombinant fast-acting variant insulin lispro, proline B28 has been replaced with lysine, which destabilizes the interaction of the two monomers.
In another fast-acting variant, insulin aspart, proline 28 is replaced with aspartic acid, which also perturbs this interaction and additionally creates electrostatic repulsion with the opposite glutamate.
| 14.7 |
The Viadur® implant |
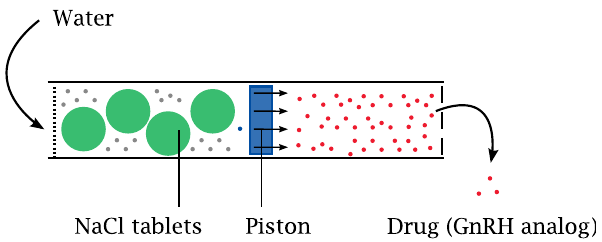
The Viadur® implant is a device for the long-term delivery of leuprolide acetate, an analogue of gonadotropin-releasing hormone (GnRH), the hypothalamic hormone that controls the release of FSH and LH from the anterior pituitary and, through these, of androgens.111
The device can sustain the release of the cargo peptide for many months; this is achieved with an “osmotic engine”. A piston inside a cylinder separates two compartments, one of which contains salt, and the other the peptide. Both ends of the cylinder are capped with membranes.
The piston moves when water enters the compartment with the salt and dissolves it. This water uptake is osmotically driven, and its rate is controlled by the permeability of the membrane delimiting this compartment. Driven by the water, the piston compresses the opposite reservoir, from which it thus expels the peptide.